Cellular Biology Techniques
Media and Solutions Required for Routine ES Cell CultureMedia used to prepare 100 mL medium:
 |
- Dulbecco’s Modification of Eagles Medium (DMEM)
- 1X without L-glutamine with 4.5 g/L glucose. Store at 4 °C
- Cytosystems 500 mL Cat. No. 11.016.0500V
- Nonessential amino acids 100X
- Cytosystems 100 mL Cat No. 21-145-0100V. Store at 4 °C
- Penicillin/streptomycin (5000 1U/mL, 5000 µg/mL)
- Cytosystems 100 mL Cat No. 21-140-0100V. Store at –20 °C
- Fetal calf serum (FCS)
- FCS
needs to be tested for the ability to support growth of ES Cells. Serum
should be stored frozen and heat-inactivated before use by heating at 56
°
for
30 mins. It may be stored at 4 °C following inactivation. The heat
inactivation removes complement activity which, along with natural
heterophile
antibodies, may be toxic for ES cells.
- 100X nucleoside stock
- To prepare 100 mL (100X)

- Add
to 100 mL Travenol water and dissolve by warming to 37°C.
Filtersterilize
and
aliquot while warm. Store at 4°C for months. The nucleosides
will
come out of solution. Warm to 37°C before use to resolubilize. Solution
seems
to be stable at 4°C.
- Other solutions required.
- 1X Dulbecco’s phosphate-buffered salt solution (PBS) without calcium and magnesium.
Cytosystems 100 mL
500 mL11.075.0100V
11.075.0500V
- 1X Dulbecco’s phosphate-buffered salt solution (PBS) without calcium and magnesium.
- Trypsin 2.5% Cytosystems 100 mL 21-159-0100V
- Trypsin/EDTA (1:250) Cytosystems (0.05% Trypsin) 100 mL 21-160-0100V
- Solutions to be made up
- PBS/EGTA (0.5 mM)
Add 1 mL of 0.05M stock EGTA (Sigma E4378) to 100 mL of PBS, to produce a final concentration of 0.5 mM.
- Stock EGTA in H2O 0.05M add concentrated NaOH to dissolve
- Trypsin 0.25%.
- Add 1.6 mL 2.5% Trypsin to 20 mL Cytosystems Trypsin/EDTA 0.05%.
- 0.1% gelatin in PBS
- Add 0.1 gm Gelatin Sigma G-1890 and autoclave to 100 mL PBS
- 0.1M 2-mercaptoethanol (§ME) sigma M-6350.
- Add 0.1 mL §ME (14.4M) to 14.3 mL PBS and filter through a 0.2 µM
- ACRODISC. Store at –20 °C for up to 1 month.
- LIF (if no feeders are used)
- LIF ESGRO AMRAD (murine Lif in PBS/BSA solution) 1 mL ampoule
- 107 U/mL. Dilute 1/100 in DMEM 10% FCS, aliquot into 10 × 10 mL
tubes, store at –20 °C until use.
- 107 U/mL. Dilute 1/100 = 105 U/mL. (100X conc)
- Dilute 1/100 for use = 1000 U/mL.
Cells are normally passaged every 2-3 days; this is important to avoid differentiation.
Signs of differentiation are:
- Colonies are surrounded by flattened, differentiated cells.
- Large colonies with necrotic centers, these appear as cells with defined
boundaries.
- Colonies appear as individual cells rather than as a syncial mass.
- Colonies are more “rounded” than “flat”, they also have a clearly defined boundary. Worse than this, they have formed free-floating embryoid bodies.
- All reagents are warmed to 37 °C.
Medium: PBS, PBS/EGTA, TRYPSIN/EDTA
- Remove medium.
- Wash with PBS (5 mL/25 cm2 flask, 10 mL/80 cm2 flask), aspirate.
- Wash with PBS/EGTA, aspirate.
- Place flask on 37 °C warming tray for approx. 1 min or until individual
cells can be seen in colonies.
- Add 0.5 mL (25 cm2), 1 mL (80 cm2) TRYPSIN/EDTA, and rock flask backwards and forwards until colonies float off; this should take ~1 min.
- Using a 1-mL pipette, pipette up and down, (avoid making bubbles as these kill the cells), for approx. 1 min. Check that all colonies have been dispersed and that a single-cell suspension has been achieved. Don’t leave cells in TRYPSIN/EDTA for longer than 3 mins, as it is quite toxic.
- Neutralize trypsin by adding an equal volume of medium, mix by gentle
pipetting.
- Aspirate media from feeder flask, as this is different media, to ES cell
media. Seed feeder* flasks with an aliquot of cell suspension. A 1:10
and
1:20
split is appropriate for a well-growing culture with medium–large
colonies that are not touching each other but are reasonably close
together.
*Feeder flasks contain Mitomycin C-treated (see protocol) Primary Mouse
Embryo Fibroblasts (PMEFs) at a concentration of 0.3 × 106/25 cm2 flask,
1 × 106/80 cm2 flask, or Mitomycin C-treated STO cells at a concentration
of 1.25 × 106/25 cm2 flask× 106/80 cm2 flask. STO cells are smaller
than PMEFs.
If cells are to be grown in the presence of LIF only, i.e., no feeder layer, flasks or plates must be treated with 0.1% gelatin in PBS at 37 °C for at least 1–2 hrs. This is removed before the medium is added..
- You will
need a 13.5-day pregnant mouse (we use MTK NEO inbred white
mice)
- 2 sets of sterile instruments, one containing a pair of curved forceps and
a pair of iris scissors
One containing 2 pairs of curved forceps, 1 pair of iris scissors, and a #3 size scalpel handle
- Phosphate buffer saline (PBS)
- Sterile medium-size petri dishes (tissue culture standard)
- 18-gauge needle
- Luer lock syringe (about 6 cc should suffice)
- #11 size flat-edged scaple blade
- Trypsin/EDTA
- Dulbecco’s Modification of Eagles Medium (with 10% fetal calf serum, 1%
penicillin/streptomycin, 1% L-glutamine, 0.2% 0.1 m BME)
- Large flasks (tissue-cultured standard, about 154 cm2 area)
- Class II Laminar Flow hood.
Using sterile forceps and iris scissors, dissect out the uterus, taking care not to touch the fur or the benchcote with the uterus or instruments. Place the uterus into a petri dish of sterile PBS and swirl around to remove blood. Transfer the uterus to a second petri dish of sterile PBS and move the dish to the hood.
Using the second set of sterile instruments and a fresh sterile petri dish, isolate the embryos. Be sure to remove the placement and embryonic sacs. Using the scalpel handle with the #11 blade on it, cut off the embryo’s head and scoop out the liver with a pair of forceps. The head and forelimb should be cut off as shown below.
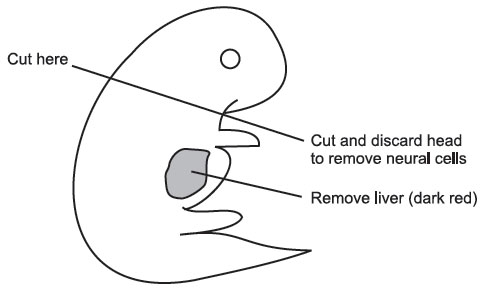 |
| Figure 14 |
Discard the head and liver and leave the bodies in fresh PBS in a fresh petri dish. Take a 6-cc luer lock syringe with an 18-gauge needle attached and remove the plunger. Keep the plunger sterile. Drop the embryo bodies inside the syringe and add 3 mL trypsin/EDTA. Put plunger back in syringe and squirt contents of syringe into a large tissue culture flask. Place the flask onto a warming tray (37 °C) for 2–3 minutes. Then place it back in the hood and add 20 mL DMEM. When adding the DMEM, try to wash any tissue off the walls
up the tissue. Transfer flask to an incubator at 37 °C with 5% CO2. Do not put less than 7 embryos in one large flask.
Important: Loosen the lid of the flask in incubator to allow gas exchange in the medium. This is the primary isolation or passage one. PMEFs should attach and begin to divide in 1–3 days. During this time, do not disturb, so as to allow PMEFs to settle and attach. After 2 days, change the medium. It will be very acidic. After 3–4 days, the culture will need splitting. Remove media and gently wash the monolayer with 2X 10 mL PBS. Add 2 mL trypsin EDTA and split 1:4. After a further 2–4 days, the culture will be ready for freezing.
The number obtained from each flask will be between 5–10 × 106 cells. Freeze cells in 10% DMSO at 3X 106/ampule.
When recovering the cells from LN2, put all the cells into a medium flask. When confluent these are split into 1 medium and 1 large flask (1:3). The large flask can be treated with mitomycin C and the medium flask split again. Do not passage beyond P6.
Media for Embryo Culture and Manipulation
M16 Medium (Protocol obtained from Karen Austen-Reed from SS Tan Laboratory, Anatomy Department)–
For oocyte maturation and routine culture of mouse embryos, M16 culture medium is used. This medium is unable to maintain its own pH, and must therefore by used in conjuction with an incubator buffered with 5% CO2. The CO2 maintains the required pH level of the medium.
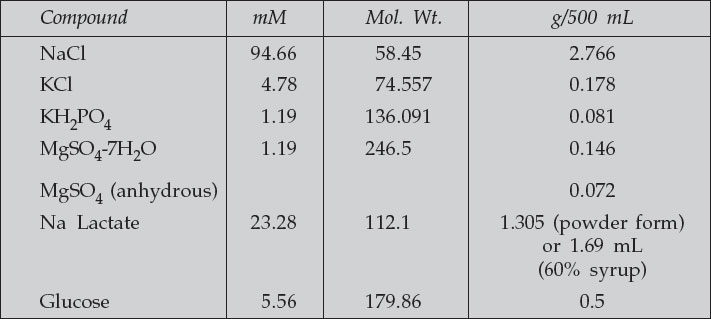 |
- Penicillin/streptomycin—use at 1/100 (Tissue culture Pen/Strep)
- Phenol red 0.005
- NaHCO3 25.0 84.02 1.051
- C Na pyruvate 0.33 110.0 0.018
- D CaCl2-2H2O 1.71 147.2 0.126
- BSA (bovine serum albumin) 4 mg/mL
- Weigh out all of stock “A” (except Pen/Strep and Na lactate) into a
measuring cylinder and make up to 90 mL with “Travenol” water. Add
Pen/Strep to cylinder and then Na lactate (Note: the Na lactate is quite
viscous—by heating it up to ~37 °C prior to use, it can be more easily
and
accurately pipetted). Make up to 100 mL with “Travenol” water and pour
into a
500-mL bottle.
- Weigh out stock “B” (Phenol red, NaHCO3)
components into a measuring
cylinder. Make up to 100 mL with “Travenol” and pour into a 500-mL
bottle. Weigh stock “C” (Na Pyruvate) into measuring cylinders, make up
to
100 mL with “Travenol” water and pour it into a 500-mL bottle. Weigh
stock
“D” (CaCl2-2H2O) into a measuring cylinder, make
up to 100 mL
with
“Travenol” water, and pour it into a 500-mL bottle. Add about 100
mL of
“Travenol” water to bring the total volume to 500 mL. Be sure that
the
components of each stock have dissolved properly before adding them
to
the 500-mL bottle. Also be sure to add stocks in their alphabetical
order
to
avoid precipitation of some ingredients.
- Make 50-mL aliquots of this solution and freeze them. Use one 50-mL
aliquot at a time by aliquoting it into tubes of 9 mL each, then
refrigerate
until
use. The osmolarity should be 288-292 m osmol.
Before use, lightly gas with CO2, then add 1 mL of FCS (fetal calf serum) to a 9-mL aliquot of M16. Then sprinkle 36 mg of BSA (4 mg/mL M16) on top and allow to dissolve—do not shake up or stir. Then Filter-sterilize this mix using a 12-mL syringe with an acrodisc.
- This medium is used for manipulations that are performed on the mouse
embryos while out of the incubator. Since there is not 5% CO2 present to
help
maintain the pH, this medium contains HEPES to keep the pH
constant.
- Ingredient % of final volume
- 1 X DMEM with 20 mM HEPES buffer 80.8%
- Penicillin/streptomycin (5000 i.u/mL/5000 µg/mL) 1%
- L-glutamine (200 mM, 100X) 1%
- Nonessential amino acid (100X) 1%
- Nucleosides (100X) 1%
- β-mercaptoethanol (0.1M) 0.2%
- Fetal calf serum 15%
- This
recipe is made up fresh each week (the same stocks of ingredients
may
be used). An alternative to DMEM with HEPES is M2, the recipe for
which
can be found in Bridgette Hogan’s Book, “Manipulating the Mouse
Embryo”.
- Before you start, have ready the following:
- Sterilize 2 pairs of curved forceps, 1 pair of iris scissors, and 2 pairs of fine watchmaker forceps.
- A 6-mL syringe filled with DMEM with HEPES, with an 18-gauge needle attached. To the needle attach a 20-cm length of clear vinyl tube, and into the end of this tubing insert a sharp flusher.
- At 2.5 days pc (post-coitus), the morulae are present in the oviducts. For this reason, it is only necessary to remove the oviducts from the mouse.
- Kill
the 2.5-days pregnant mouse and lay it on its back on benchcloth or
absorbent paper. Swab its belly with ethanol (70%) and nick the skin
with
a
pair of scissors. Pull the skin back over the head and toward the tail.
Cut
and
pull back the body wall to expose the contents of the abdomen. Push
aside
the guts to expose the reproductive tract. Using a pair of curved
forceps, grasp the uterus just below the oviduct and carefully separate
the
oviduct from the ovary using the iris scissors. Cut through the uterus
just
below
the uterotubal junction and place the oviduct in a petri dish.
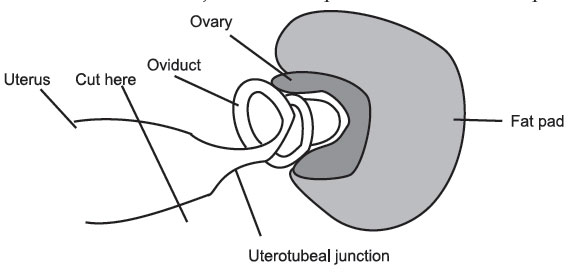
Figure 15
- Transfer
the dish to the stage of a stereo dissecting microscope. Using the
watchmaker forceps, manipulate the oviduct so that you have the end of
the
oviduct (close as possible to the uterotubal junction) between the
forceps
and
insert the flusher so that it points away from the uterotubal junction.
Be
careful not to pierce through both sides of the oviduct tube. Keeping
the
flusher inserted into the oviduct, pick up the syringe in the other
hand,
and
give a couple of short, sharp squeezes on the plunger. This should
flush
all morulae from the oviduct. Sometimes, if no morulae are flushed
through, it is hard to tell whether the oviduct has been correctly flushed. If there is much debris floating around, then the oviduct has been properly flushed. The morulae are then collected using a mouth pipette and deposited into a petri dish in microdrops of M16 medium. These drops are covered with fluid 200 (Dow Corning, viscosity 50CS) and placed in a 37 °C incubator buffered with 5% CO2.
- Blastocyst transfer is usually performed 24 hours after aggregation when
the
morulae have become expanded blastocysts, on the same day as the
injection. A little time is given between injection and transfer to
allow
blastocysts to re-expand.
- Careful selection of the recipient is most important, since the pups are
the
end
result of a lot of hard work. Two strains of mice are used. RB Swiss
and
(CBA*C57BL6/J)f1. RB Swiss are quiet and make excellent mothers but
they
become overweight quickly and do not take anesthesia well. CBA*C57
mice
are hardy and display hybrid vigor. They do not carry excess weight
and
go under anesthetic well. This strain can be very nervous when
housed separately, which could be dangerous to their young. They are
most
suitable if a young RB Swiss is placed into the cage as a companion
that
can be removed as soon as the pups are 7 days old. By this age,
destruction of the litter is very unlikely. If the CBA mother is to be
housed
alone, she must not be disturbed for 10 days.
Prior to surgery, sterilize the following:
- 3 pairs of curved forceps
- 2 pairs of iris scissors
- 1 pair suture clamps
- 1 serafin clip
- Sterile suture with needle attached (small—for mouse surgery)
- Michelle clips (small size)
- Michelle clip applicators
- 1 mouth pipette and flame-polished transfer pipette
- You will also need an anaesthetic. Rompun/Ketavet is found to be quite effective. To make up 10 mL:
- 0.5 mL 2% Rompun (20 mg/mL Xylazine)
- 0.5 mL 100 mg/mL Ketavet 100—Delta Veterinary Lab, 8 Rosemead Rd Hornsby NSW 2077
- Make up to 10 mL with PBS
- The dosage is 0.02 mL/g body weight.
- Store wrapped in tin foil at 4 °C
- Shake well before use, as it tends to separate in the fridge
- Select a mouse that is 2.5 days pseudopregnant and weigh. Do not use
anything lighter than 25 g or anything heavier than 30 g. Underweight
mice
tend to reabsorb the embryos, as they are not physically ready to
support a pregnancy. Overweight mice make surgery difficult since the
absorption of anesthetic into the fat reduces the potency of the
anesthetic.
Also,
the presence of fat means the presence of blood vessels, and cutting
through all the extra fat causes a lot of unnecessary bleeding. This
makes
it
difficult to see what you are doing and may also clog up the tip of your
transfer pipette.
- Anesthetize the mouse with Rompun/Ketavet, administered
intraperitoneally.
After
administering the anesthetic, put the mouse back into the box
from
which it came. The mouse will be more relaxed when placed in a
familiar environment and the anesthetic will act more quickly than it
would
on a distressed mouse.
- To
check that the mouse is fully anesthetized, press or squeeze the pads
of
the feet. If the mouse can feel this, it will try to withdraw its leg
from
your
grasp. Do not commence surgery until there is no reflex reaction to
this
test.
- Take the anesthetized mouse and shave its lower back. Lay the mouse on
its
belly on a petri dish lid, taking care to keep the airway clear by
resting
the
teeth on the edge of the petri dish. This makes it easier to move the
mouse
around without having to actually touch it. Swab the shaven area
with
hibitane or 70% ethanol.
- Instruments should have been laid out. Use 1 pair of iris scissors and 1 pair of forceps for cutting the skin—call these “outside” instruments. Use 1 pair of iris scissors and 2 pair of forceps for working inside the mouse— called the “inside” instruments.
- Using the outside forceps and scissors, make a small cut (about 1-cm long) along the dorsal midline of the lower back. Through the shaven, moistened skin, it is fairly easy to see blood vessels. Try to avoid these vessels when making the incision (see below).
- Using
a pair of outside forceps and inside forceps, pick up the skin and
separate it from the body wall. Cut the body wall as indicated about
5-mm
long, avoiding blood vessels.
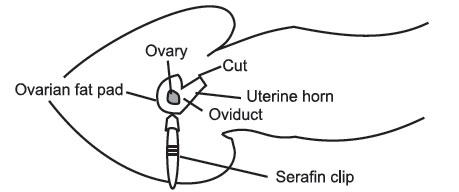
- If
the cut has been made in the right place, the ovarian fat pad is easily
visible. If not, the fat pad can be located by lifting the edge of the
body
wall
and scouting around with the other pair of inside forceps. Once you
have
located the fat pad attach the serafin clip to it, taking care not to
clip
the
ovary. It is important not to damage the ovary, as it is responsible for
hormone production throughout pregnancy. Gently ease the ovary, oviduct,
and
part of the uterus out through the incision in the body wall. Do not
Pull. A traumatized uterus will contract and move quite violently, making
surgery difficult, and may cause expulsion of the transferred blastocysts. When the tip of the uterus is visible, rest the serafin clip across the mouse’s back to hold the uterus in place. If the uterus or uterine horn continually slip back into the cavity, it may be necessary to gently lie the mouse on the side, being careful not to block the airway.
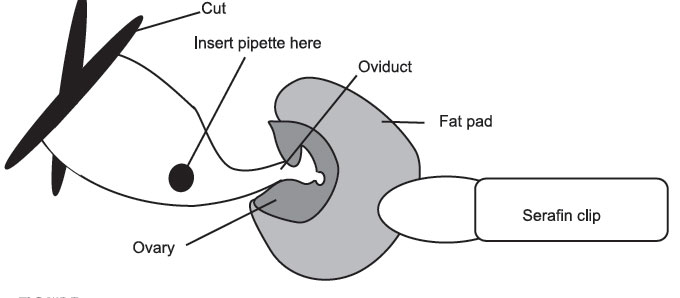
Figure 17
- The
transfer pipette should now be loaded. Five or six embryos must be
transferred to each horn; any less than this and the chances of a
pregnancy
resulting are serverely reduced. It may be loaded in such a way to suit
yourself, but this is a method that is popular. Take up a minute amount
of
DMEM with Hepes in the very tip of the transfer pipette, then make a
small
bubble by taking up a little air. Then take up some more medium—
roughly the same volume as you hope to transfer the blastocysts in. Take
up
another bubble, the same size as before. Then take up your blastocysts
in the smallest possible volume of medium, lining them up side by side in the transfer pipette. This is how your transfer pipette should look when loaded.
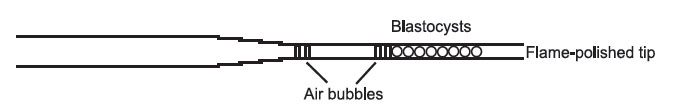
Figure 18
- This
will take some practice. Make sure that you are competent at loading
a
transfer pipette before any attempt at a blastocyst transfer. During
surgery
is
not the time to learn how to load a pipette. If it is likely to take you
more
than a
few minutes to load the transfer pipette, then do not expose the
uterus until the pipette has been loaded. This prevents drying out and
further trauma to the uterus. Alternatively, the uterus, ovary, etc.,
may be
moistened repeatedly with a sterile cotton bud and saline.
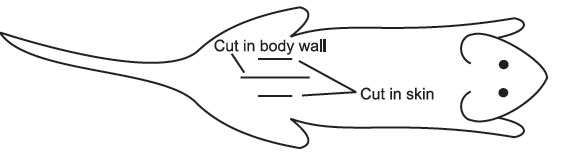
Figure 19
- Once the pipette is loaded and the uterus positioned, move the petri dish lid supporting the mouse to the microscope and turn on the overhead light source. Once the lights and focus have been adjusted and the mouse positioned to suit yourself, gently grasp the top of the uterine horn with a pair of inside forceps. While still holding the horn with one hand, use the other hand to gently insert a 26-gauge hypodermic needle through the uterine wall (close to the oviduct) and into the lumen. Choose an area of the uterus that is relatively devoid of blood vessels, as blood will clot in the tip of your pipette and block it. Remove the needle carefully (so as not to lose sight of the hole), without averting your eyes, pick up the loaded transfer pipette. Gently insert the transfer pipette about 3 mm into the uterine lumen. Gently blow the blastocysts into the uterus, using the air bubbles in your transfer pipette to monitor the transfer. Be careful not to blow any air into the uterus. Once transfer is complete, quickly rinse out transfer pipette in some HEPES buffer medium (M2) and check to see if there were any blastocysts stuck in the transfer pipette. If there were, transfer these blastocysts again.
- With the transfer complete, the serafin clip can now be removed and the
uterus gently eased back into the body. Do not touch the uterus, but
ease
it
back by the edges of the incision in the body wall and allowing the
uterus
to
fall back in, without actually handling it. This procedure is then
repeated
on
the other uterine horn. The incision in the body wall is not sutured.
The
skin
is closed with Michelle clips—2 per incision is usually sufficient.
Michelle clips are used on the skin instead of sutures, because the mice
will
chew at the suture thread and effectively open their wounds up.
- Once surgery is complete, the mouse is placed in a box of clean
autoclaved
sawdust. Under anesthetic, mammals are unable to retain heat as
effectively
as
when conscious. For this reason, and also because the animal has been
shaven, the mouse should be wrapped in a tissue to help keep it warm.
This
will also be used as bedding until the fur has grown back. It is also
very
important that the mouse be housed alone, as anesthetized animals
are
often buried by cagemates who think they are dead. All animals should
have
recovered sufficiently from the anesthetic before being returned to the
animal room and left unattended. The cage should be placed on a shelf
away
from male mice, as strange male pheromones will often cause females
to
abort. Recipient mice should be handled with care and tiptoed around
as
pregnant mice are easily upset, sometimes leading to abortion, or even
cannibalism of pups.
 |
| Figure 16 |
Stock Solutions
- M CaCl2 Merck. Filter (to remove particulates), autoclave, and store at
–20°C in aliquots.
- 2X HEBS gm/500 mL
- NaCl 8.0
- KCl 0.38
- Na2HPO4.7H2O 0.19
- Glucose 1.0
- HEPES 5.0
- Adjust pH to 7.05 ± .05 with NaOH, autoclave, and store frozen in aliquots.
- NaCl 8.0
- 1 X HEBS/15% glycerol. 50 mL 2X HEBS)
- 15 mL glycerol
- 35 mL DDW, autoclave and store at –20°C in aliquots.
- 15 mL glycerol
- Carrier DNA. Mouse liver DNA, sheared at 90 µg/mL in 0.2X SSC by
passing 2X through a 20-gauge needle; filtered through a 4.45-mm filter.
- Plate cells at 1X 106 in 10 cm dishes.
- The next day, perform transfection.
- Set up 2 rows of tubes—A & B
- In tube A place:15 ug of plasmid DNA (linearized, phenol-extracted, precipitated, and resuspended at 1 µg/mL in sterile, 0.2X SSC, 69 µL 2M CaCl2, 460 0.2X SSC.
- In tube B, place 550 µL 2X HEBS.
- Using an autopipette and a 1-mL pipette, have tube B bubbling while
slowly adding contents of tube A.
- Stand 15–20 mins, while precipitate forms, producing a milky fine pipette.
- Carefully add precipitate dropwise to a 10-cm dish of cells while maintaining
the pH of cultures.
- Leave 3–4 hrs in CO2 incubator (Can be left overnight).
- Glycerol Shock:
Aspirate medium: Wash by adding ~3 mL medium then aspirate. Add 2.5 mL 15% glycerol in HEBS.
Sit 4 mins at room temp., then aspirate. Wash with 5 mL medium per plate. Add fresh medium.
Cells are fragile and only loosely attached at this stage. Handle very gently. If pipette was left on cells overnight, do the splits the next day.
Next day
- Trypsinize to harvest cells. Recover cells in medium containing
antibiotic
and
divide directly in the ratio 3/5 and 1/20th into two 10-cm plates,
respectively.
- Feed it every 2–3 days with a medium containing antibiotic (once a week
when most cells have been killed).
Materials
- Sterile technique
- Autoclave 1 X PBS
- Dissection tools (watchmaker forceps X 2–3, surgical scissors)
- Sterile dishes (10 cm)
- Prepare 6 well dishes to culture the embryo cell culture and mark the well.
- Prepare syringe contain required amount (300 µL) of trypsin and keep in
- hood.
- Stopcock 3-way transfusion
- Kill mice.
- Open abdomen and remove the both side of the uterus—sterile from now!
- Open uterus and separate embryo (include decidua).
- Take single embryo into fresh dish, open decidua, open and remove york
sack
for PCR genotype (soak in PBS of the 6 well dishes, wash twice, and
dip
into DNA buffer).
- Rinse the forceps, take the embryo into a fresh syringe, carry the
syringe
contain the embryo into a tissue culture hood. Place on a 3-way stopcock
and
add on a syringe containing 300 µL trypsin on the other side. Start
timing before pushing the syringe. Push the syringe from one side at a
time,
carefully close the stopcock bit by bit (not too much as the cells will
be
lysed). This requires about 20 seconds (shorter is better), and place
the
syringe in one well, then place the dish on a warm plate, up to 3
minutes
in
total.
- Rinse the syringe with 1 mL media and add total media of 3–3.5 mL. Do
not
use a pipette. Take up and push down the culture, since the embryo
cells
tend to be happier when the cells are aggregated.
Incubate the culture at 37°C, 5% CO2 for 3–5 days until the
cells reach
confluence. Transfer the cell into a 10-cm dish by 1 mL trypsin and 8–10
Materials
- DNA buffer:1 X PCR buffer
- TISSUE CULTURE TECHNIQUES 573
- 0.45% NP-40
- 0.45% Tween-20
- Proteinase K 100 mg/mL
- In a PCR tube containing 50 µL DNA buffer.
- Dig washed york sack into tube, spin down.
- Incubate at 50°C for 30 minutes.
- Boil the lysate for 5 minutes, spin down.
- Take 5 µL for PCR.
- 1 X PCR buffer
- 2 mM MgCl2
- 100 nM dNTPs
- 0.25 mM
- 0.05 mM
- 0.2 mM
- a c 400bp (targeting region)
- a b 200bp (wild type)
- 2.5 unite/100 µL Taq DNA polymerase
- Initial denaturing by 94°C for 4 minutes
- 28 cycles of 94°C for 1 minute
- 60°C for 1 minute
- 72°C for 1 minute
- Final extension by 72°C for 10 minutes.
Support our developers




