Tissue Culture Methods
Types of Cells Grown in CultureTissue culture is a term that refers to both organ culture and cell culture. Cell cultures are derived from either primary tissue explants or cell suspensions. Primary cell cultures typically will have a finite life span in culture, whereas continuous cell lines are, by definition, abnormal and are often transformed cell lines.
Work Area and Equipment
- Laminar Flow Hoods: There are 2 types of laminar flow hoods, vertical and
horizontal, and both types of hoods are available in the microbiology
laboratory. The vertical hood is best for working with hazardous organisms,
since the aerosols that are generated in the hood are filtered out before they
are released into the surrounding environment. Horizontal hoods are
designed so that the air flows directly at the operator, hence, they are not
useful for working with hazardous organisms, but are the best protection
for your cultures.
Both types of hoods have continuous displacement of air that passes through a HEPA (high efficiency particle) filter that removes particulates from the air. The hoods are equipped with a shortwave UV light that can be turned on for a few minutes to sterilize the surfaces of the hood, but be aware that only exposed surfaces will be accessible to the UV light. Do not put your hands or face near the hood when the UV light is on, as the shortwave light can cause skin and eye damage. The hoods should be turned on about 10–20 minutes before being used. Wipe down all surfaces with ethanol before and after each use. - Microscopes: Inverted phase contrast microscopes are used for visualizing the cells. Microscopes should be kept covered and the lights turned down when not in use. Before using the microscope or whenever an objective is changed, check that the phase rings are aligned.
- CO2 Incubators: The cells are grown in an atmosphere of 5%–10% CO2, because the medium used is buffered with sodium bicarbonate/carbonic acid and the pH must be strictly maintained. Culture flasks should have loosened caps to allow for sufficient gas exchange. The humidity must also be maintained for those cells growing in tissue culture dishes, so a pan of water is kept filled at all times.
- Preservation: Cells are stored in liquid nitrogen.
- Vessels: Anchorage-dependent cells require a nontoxic, biologically inert, and optically transparent surface that will allow cells to attach and allow movement for growth. The most convenient vessels are specially-treated polystyrene plastic that are supplied sterile and are disposable. These include petri dishes, multiwell plates, microtiter plates, roller bottles, and screwcap flasks.
Preservation and Storage
Liquid N2 is used to preserve tissue culture cells, either in the liquid phase or in the vapor phase. Freezing can be lethal to cells due to the effects of damage by ice crystals, alterations in the concentration of electrolytes, dehydration, and changes in pH. To minimize the effects of freezing, several precautions are taken. First, a cryoprotective agent that lowers the freezing point, such as glycerol or DMSO, is added.
The freezing medium is typically 90% serum, 10% DMSO. In addition, it is best to use healthy cells that are growing in log phase and to replace the medium 24 hours before freezing. Also, the cells are slowly cooled from room temperature to –80°C to allow the water to move out of the cells before it freezes. The optimal rate of cooling is 1°C–3°C per minute. Some labs have fancy freezing chambers to regulate the freezing at the optimal rate by periodically pulsing in liquid nitrogen.
The tubes filled with 200 mL of isopropanol at room temperature and the freezing vials containing the cells are placed in the container. The container is placed in the −80°C freezer. The effect of the isopropanol is to allow the tubes to come to the temperature of the freezer slowly, at about 1°C per minute.
Once the container has reached −80°C the vials are removed and immediately placed in the liquid nitrogen storage tank. Cells are stored at liquid nitrogen temperatures because the growth of ice crystals is retarded below −130°C.
Maintenance
Cultures should be examined daily, observing the morphology, the color of the medium, and the density of the cells. A tissue culture log should be maintained. The log should contain the name of the cell line, the medium components, and any alterations to the standard medium, the dates on which the cells were split and/or fed, a calculation of the doubling time of the culture (this should be done at least once during the semester), and any observations relative to the morphology, etc.
- Growth Pattern: Cells will initially go through a quiescent or lag phase that depends on the cell type, the seeding density, the media components, and previous handling. The cells will then go into exponential growth where they have the highest metabolic activity. The cells will then enter into stationary phase where the number of cells is constant. This is characteristic of a confluent population (where all growth surfaces are covered).
- Harvesting: Cells are harvested when the cells have reached a population
density that suppresses growth. Ideally, cells are harvested when they are
in a semiconfluent state and are still in log phase. Cells that are not
passaged and are allowed to grow to a confluent state can sometime lag for a long period of time, and some may never recover. It is also essential to keep your cells as happy as possible to maximize the efficiency of transformation. Most cells are passaged (or at least fed) 3 times a week.
- Suspension cultures: Suspension cultures are fed by dilution into a fresh medium.
- Adherent cultures: Adherent cultures that do not need to be divided can simply be fed by removing the old medium and replacing it with fresh medium. When the cells become semiconfluent, several methods are used to remove the cells from the growing surface so that they can be diluted:
Proteolytic enzymes:. Trypsin, collagenase, or pronase, usually in combination with EDTA, causes cells to detach from the growth surface. This method is fast and reliable but can damage the cell surface by digesting exposed cell surface proteins. The proteolysis reaction can be quickly terminated by the addition of complete medium containing serum.
EDTA: EDTA alone can also be used to detach cells and seems to be gentler on the cells than trypsin.
- Visually inspect daily.
- Release cells from monolayer surface
- Wash once with a buffer solution.
- Treat with dissociating agent.
- Observe cells under the microscope. Incubate until cells become rounded and loosen when flask is gently tapped with the side of the hand.
- Transfer cells to a culture tube and dilute with medium containing serum.
- Spin down cells, remove supernatant, and replace with a fresh medium. Count the cells in a hemocytometer, and dilute as appropriate into a fresh medium.
- Media and Growth Requirements
- Physiological parameters
- emperature –37°C for cells from homeotherms.
- pH–7.2–7.5 and osmolality of medium must be maintained.
- Humidity is required.
- Gas phase-bicarbonate concentration and CO2 tension in equilibrium.
- Visible light can have an adverse effect on cells; light-induced
production of toxic compounds can occur in some media; cells
should be cultured in the dark and exposed to room light as little
as possible.
- Medium requirements: (often empirical)
- Bulk ions—Na, K, Ca, Mg, Cl, P, Bicarb, or CO2.
- Trace elements—iron, zinc, selenium.
- Sugars—glucose is the most common.
- Amino acids—13 essential ones.
- Vitamins—B, etc.
- Choline—inositol.
- Serum contains a large number of growth-promoting activities, such as buffering toxic nutrients by binding them, neutralizes trypsin and other proteases, has undefined effects on the interaction between cells and substrate, and contains peptide hormones or hormonelike growth factors that promote healthy growth.
- Antibiotics, although not required for cell growth, are often used to
control the growth of bacterial and fungal contaminants.
For our purposes, we will use the following media components:
Basal medium—IMDM
Serum—10% fetal calf - Glutamine—1%—an essential amino acid that tends to be unstable— it is typically stored frozen and added separately; its half-life in medium at 4°C is 3 weeks, at 37°C, 1 week.
- Antibiotic/antimycotic—1% (streptomycin, amphotericin B, penicillin; spectrum: bacteria, fungi and yeast).
- Feeding—2–3 times/week.
- Measurement of growth and viability. The viability of cells can be observed visually using an inverted phase contrast microscope. Live cells are phase bright; suspension cells are typically rounded and somewhat symmetrical; adherent cells will form projections when they attach to the growth surface. Viability can also be assessed using the vital dye, trypan blue, which is excluded by live cells but accumulates in dead cells. Cell numbers are determined using a hemocytometer.
- Physiological parameters
Safety Considerations
Assume all cultures are hazardous since they may harbor latent viruses or other organisms that are uncharacterized. The following safety precautions should also be observed:
- Pipetting: use pipette aids to prevent ingestion and keep aerosols down to a minimum.
- No eating, drinking, or smoking.
- Wash hands after handling cultures and before leaving the lab.
- Decontaminate work surfaces with disinfectant (before and after).
- Autoclave all waste.
- Use biological safety cabinet (laminar flow hood) when working with hazardous organisms. The cabinet protects worker by preventing airborne cells and viruses released during experimental activity from escaping the cabinet; there is an air barrier at the front opening and exhaust air is filtered with a HEPA filter.
- Make sure the cabinet is not overloaded and leave exhaust grills in the front and the back clear (helps to maintain a uniform airflow).
- Use aseptic technique.
- Dispose of all liquid waste after each experiment and treat with bleach.
Tissue Culture Methods
Each student should maintain his or her own cells throughout the course of the experiment. These cells should be monitored daily for morphology and growth characteristics, fed every 2 to 3 days, and subcultured when necessary. A minimum of two 25-cm2 flasks should be carried for each cell line; these cells should be expanded as necessary for the transfection experiments. Each time the cells are subcultured, a viable cell count should be done, the subculture dilutions should be noted, and after several passages, a doubling time determined. As soon as you have enough cells, several vials should be frozen away and stored in liquid N2. One vial from each freeze down should be thawed 1–2 weeks after freezing to check for viability. These frozen stocks will prove to be vital if any of your cultures become contaminated.
Procedures
- Media preparation: Each student will be responsible for maintaining his or
her own stock of cell culture media; the particular type of media, the sera
type, and concentration, and other supplements will depend on the cell
line. Do not share media with your partner (or anyone else), because if a
culture or a bottle of media gets contaminated, you have no back-up. Most
of the media components will be purchased prepared and sterile. In general,
all you need to do is sterilely combine several sterile solutions. To test
for sterility after adding all components, pipette several mL from each
media bottle into a small sterile petri dish or culture tube and incubate at
37°C for several days. Use only media that has been sterility-tested. For
this reason, you must anticipate your culture needs in advance so you can
prepare the reagents necessary. But, please, try not to waste media. Anticipate
your needs but don’t make more than you need. Tissue culture reagents
are very expensive; for example, bovine fetal calf serum cost ~$200
per 500 mL. Some cell culture additives will be provided in a powdered
form. These should be reconstituted to the appropriate concentration with
double-distilled water (or medium, as appropriate) and filtered (in a sterile
hood) through a 0–22 µm filter.
All media preparation and other cell culture work must be performed in a laminar flow hood. Before beginning your work, turn on the blower for several minutes, wipe down all surfaces with 70% ethanol, and ethanolwash your clean hands. Use only sterile pipettes, disposable test tubes, and autoclaved pipette tips for cell culture. All culture vessels, test tubes, pipette tip boxes, stocks of sterile eppendorfs, etc., should be opened only in the laminar flow hood. If something is opened elsewhere in the lab by accident, you can probably assume it’s contaminated. If something does become contaminated, immediately discard the contaminated materials into the biohazard container and notify the instructor. - Growth and morphology: Visually inspect cells frequently. Cell culture is sometimes more an art than a science. Get to know what makes your cells happy. Frequent feeding is important for maintaining the pH balance of the medium and for eliminating waste products. Cells do not typically like to be too confluent, so they should be subcultured when they are in a semiconfluent state. In general, mammalian cells should be handled gently. They should not be vortexed, vigorously pipetted or centrifuged at greater than 1500 g.
- Cell feeding: Use prewarmed media and have cells out of the incubator for
as little time as possible. Use 10–15 mL for T-25s, 25–35 mL for T-75s, and
50–60 mL for T-150s.
- Suspension cultures: Feeding and subculturing suspension cultures are done simultaneously. About every 2–3 days, dilute the cells into fresh media. The dilution you use will depend on the density of the cells and how quickly they divide, which only you can determine. Typically 1:4 to 1:20 dilutions are appropriate for most cell lines.
- Adherent cells: About every 2–3 days, pour off old media from culture flasks and replace with fresh media. Subculture cells as described below before confluency is reached.
- Subculturing adherent cells: When adherent cells become semiconfluent,
subculture using 2 mm EDTA or trypsin/EDTA.
Trypsin-EDTA:
- Remove medium from culture dish and wash cells in a balanced salt solution without Ca++ or Mg++. Remove the wash solution.
- Add enough trypsin-EDTA solution to cover the bottom of the culture vessel and then pour off the excess.
- Place culture in the 37°C incubator for 2 minutes.
- Monitor cells under microscope. Cells are beginning to detach when they appear rounded.
- As soon as cells are in suspension, immediately add the culture medium containing serum. Wash cells once with serum-containing medium and dilute as appropriate (generally 4- to 20-fold).
- Prepare a 2 mm EDTA solution in a balanced salt solution (i.e., PBS without Ca++ or Mg++).
- Remove the medium from the culture vessel by aspiration and wash the monolayer to remove all traces of serum. Remove salt solution by aspiration.
- Dispense enough EDTA solution into culture vessels to completely cover the monolayer of cells.
- The coated cells are allowed to incubate until cells detach from the surface. Progress can be checked by examination with an inverted microscope. Cells can be gently nudged by banging the side of the flask against the palm of the hand.
- Dilute cells with fresh medium and transfer to a sterile centrifuge tube.
- Spin cells down, remove supernatant, and resuspend in culture medium (or freezing medium if cells are to be frozen). Dilute as appropriate into culture flasks.
- Thawing frozen cells
- Remove cells from frozen storage and quickly thaw in a 37°C waterbath
by gently agitating vial. - As soon as the ice crystals melt, pipette gently into a culture flask containing prewarmed growth medium.
- Log out cells in the “Liquid Nitrogen Freezer Log” Book.
- Remove cells from frozen storage and quickly thaw in a 37°C waterbath
- Freezing cells
- Harvest cells as usual and wash once with complete medium.
- Resuspend cells in complete medium and determine cell count/viability.
- Centrifuge and resuspend in ice-cold freezing medium: 90% calf serum/ 10% DMSO, at 106–107; cells/mL. Keep cells on ice.
- Transfer 1-mL aliquots to freezer vials on ice.
- Place in a Mr. Frosty container that is at room temperature and has sufficient isopropanol.
- Place the Mr. Frosty in the –70°C freezer overnight. Note: Cells should be exposed to freezing medium for as little time as possible prior to freezing.
- The next day, transfer to liquid nitrogen (DON’T FORGET) and log in the “Liquid Nitrogen Freezer Log” Book.
- Viable cell counts: Use a hemocytometer to determine total cell counts and viable cell numbers.
Trypan blue is one of several stains recommended for use in dye exclusion procedures for viable cell counting. This method is based on the principle that live cells do not take up certain dyes, whereas dead cells do.
- Prepare a cell suspension, either directly from a cell culture or from a concentrated or diluted suspension (depending on the cell density) and combine 20 µL of cells with 20 µL of trypan blue suspension (0.4%). Mix thoroughly and allow to stand for 5–15 minutes.
- With the cover slip in place, transfer a small amount of trypan blue-cell suspension to both chambers of the hemocytometer by carefully touching the edge of the cover slip with the pipette tip and allowing each chamber to fill by capillary action. Do not overfill or underfill the chambers.
- Starting with 1 chamber of the hemocytometer, count all the cells in the 1-mm center square and each of the four 1-mm corner squares. Keep a separate count of viable and nonviable cells.
- If there are too many or too few cells to count, repeat the procedure, either concentrating or diluting the original suspension as appropriate.
- The circle indicates the approximate area covered at 100X microscope
magnification (10X ocular and 10X objective). Include cells on top and left,
touching the middle line. Do not count cells touching the middle line at
the bottom and right. Count 4 corner squares and the middle square in both chambers and calculate the average. - Each large square of the hemocytometer, with cover slip in place, represents
a total volume of 0.1 mm3 or 10−4 cm3. Since 1 cm3 is equivalent to
approximately 1 mL, the total number of cells per mL will be determined
using the following calculations:
Cells/mL = average cell count per square × dilution factor × 104;
Total cells = cells/mL × the original volume of fluid from which the cell
sample was removed; % Cell viability = total viable cells (unstained)/total
cells × 100.
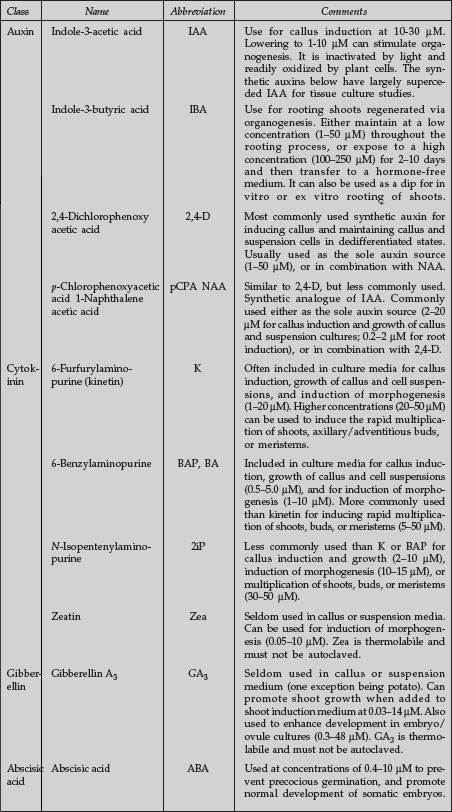 |
| Plant Growth Regulators Commonly Used in Plant Tissue Culture |
Support our developers




