Angiogenesis Assays
I. INTRODUCTIONAll healthy and pathological tissue growth requires the formation of functional blood vessels (Hanahan and Folkman, 1996). Angiogenesis, sprouting of new capillaries from the existing vessels, is the key process that contributes to both physiological and pathological neovascularization. Angiogenesis has paradoxical implications in the treatment of the most severe human diseases. In disorders such as atherosclerosis/ infarction of the heart, stroke, and peripheral occlusive limb disease, the growth of new vessels in the affected tissue is obviously beneficial for the improvement of blood circulation (Cao et al., 2003). In addition, organ transplantation and tissue regeneration need new blood vessels. However, inhibition of angiogenesis has become an important therapeutic approach for the treatment of diseases such as cancer, diabetic retinopathy, and arthritis in which angiogenesis plays an important role (Cao, 2001; Folkman, 1995). It has been reported that therapeutic angiogenesis or antiangiogenesis could delay the onset and progression of obesity (Rupnick et al., 2002). Based on their broad therapeutic implications and potential economic values, discovery and development of novel angiogenic and antiangiogenic agents have become a competitive business for pharmaceutical companies. As a result, a novel angiogenesis regulator is identified almost every week. There are at present many assay systems to be used for testing the angiogenic or antiangiogenic activity of compounds (Jain et al., 1997; Kenyon et al., 1996). The process of angiogenesis includes several critical steps: endothelial cell morphological changes, endothelial migration, endothelial proliferation, endothelial cell reorganization, and lumen formation, formation of new branches, followed by reconstitution of the basement membrane and vascular remodeling. Each of these steps can be monitored using appropriate in vitro and in vivo assay systems. This article describes in vivo and in vitro assays that are reliable, reproducible, and convenient and that are important for the characterization of compounds for use in angiogenesis-related disorders. Migration assays are described elsewhere in this volume, while this article includes the capillary endothelial cell proliferation assay and functional vessel formation assays, including the shell-less in vivo chick chorioallantoic membrane (CAM) assay and the mouse corneal angiogenesis assay.
II. MATERIALS AND INSTRUMENTS
A. Endothelial Proliferation Assay
Primary bovine capillary endothelial (BCE) cells are from Dr. Judah Folkman's laboratory at the Children's Hospital, Boston. All tissue culture plastic bottles and discs are from Falcon, Becton Dickinson. DME (low glucose) medium is from JRH Biosciences Limited (Cat. No. 2D0113). Gelatin is from Difco (Becton Dickinson, Cat. No. 214340). Recombinant human fibroblast growth factor (FGF-2) is from Scios Nova (Mountain View, CA). Bovine calf serum (BCS) is from Boule Nordic AB (Cat. No. SH300072.03). Trypsin solution (0.05%) is from Sigma (Cat. No. T9906). The Isoton phosphate-buffered saline (PBS) solution is from KEBO (Stockholm, Sweden). The Coulter counter is by Coulter, KEBO, Sweden.
B. Chick Chorioallantoic Membrane Assay
Fertilized white leghorn eggs are from OVA Production, Sörgåden, Sweden. Epigallocatechin-3-gallate is from Sigma (Cat. No. E4143). Methylcellulose is from Sigma (Cat. No.M-0262). Tissue culture plates (100 × 20 mm) are from Falcon, Becton Dickinson. The chick embryo incubator is made in Germany. The stereomicroscope is from Nikon, Japan.
C. Mouse Corneal Angiogenesis Assay
C57B16/J male and female mice are from the Microbiology and Tumor Biology Center, Karolinska Institute, Stockholm, Sweden. Vascular endothelial growth factor (VEGF) is from R&D System Inc. (Cat. No. 293-VE-050). FGF-2 is from Scios Nova. Sulcralfate is from Bukh Meditec, Vaerlose, Denmark. Hydron (type NCC) is from Interferon Sciences, Inc. (New Brunswick, NJ) (Cat. No. NCC-97001). Nylon mesh (15 x 15 mm) is from Tetko (Lancaster, NY). Methoxyflurane and proparacaine are from Ophthetic (Alcon, TX). The operation microscope is made by Zeiss, Oberkochen, Germany. Surgical blades are from Bard- Parker No. 15; Becton Dickinson (Franklin Lakes, NY). The eye examination microscope is from Nikon, Sf-2, Tokyo, Japan.
III. PROCEDURES
A. Endothelial Cell Proliferation Assay
For additional information, see Cao et al. (1996, 1997, 1999, 2001).
1. Stimulation of Endothelial Cell Proliferation
Steps
- Set a CO2 incubator to 10% CO2
- Set up tissue culture hood for at least 15 min before use.
- Coat 24-well tissue culture plates with 0.5 ml 1.5% gelatin in PBS solution (previously sterilized by autoclaving) /well for at least 1 h at 37°C or overnight at room temperature.
- Prewarm the following solution to room temperature: 1× PBS, 0.05% trypsin solution, and 10% BCSDMEM medium.
- Thaw the FGF-2 solution in ice and dilute with sterile 10% BCS-DMEM to appropriate concentrations.
- Add FGF-2 to 10% BCS-DMEM to a final concentration of 3 ng/ml.
- Remove culture medium from BCE cells (cultured in 6-well plates) and wash cells with PBS twice.
- Add 0.5 ml trypsin solution/well and expose cells to trypsin solution until they detach from the bottom.
- Once cells are detached, add immediately 5 ml/well of 10% BCS-DMEM to cells.
- Transfer cell solution into a centrifuge tube and centrifuge cells at 1500rpm for 5 min.
- Remove supernatant and resuspend cells in 10 ml 10% BCS-DMEM containing 3ng/ml FGF-2.
- Count cell numbers and seed cells at a density of 10,000 cells/well. Make sure that each sample is at least in triplicate.
- Incubate cells at 37°C and 10% CO2 for 72h.
- After 72 h, remove the culture medium and wash cells twice with PBS.
- Add 0.5ml of 0.05% trypsin solution to each well.
- When cells are detached completely, resuspend cells into a single cell solution by tituration.
- Transfer cell solution to a Coulter cup containing 10ml Isoton solution and counter cell numbers.
2. Inhibition of Endothelial Cell Proliferation
- Repeat steps 1-10 described in Section III,A,1.
- Resuspend cells in 5% BCS-DMEM in an appropriate volume.
- Seed BCE cells at a density of 10,000 cells/well in a volume of 0.5 ml of 5% BCS-DMEM.
- Add tested angiogenesis inhibitors (e.g., angiostatin or EGCG) at various concentrations and incubate the cells for 1 h at 37°C with 10% CO2
- Add FGF-2 to a final concentration of I ng/ml and incubate the cells for 72h.
- Repeat steps 14-17 described in Section III,A,1.
B. Chick Chorioallantoic Membrane Assay
For additional information, see Cao et al. (1998, 1999, 2001).
Steps
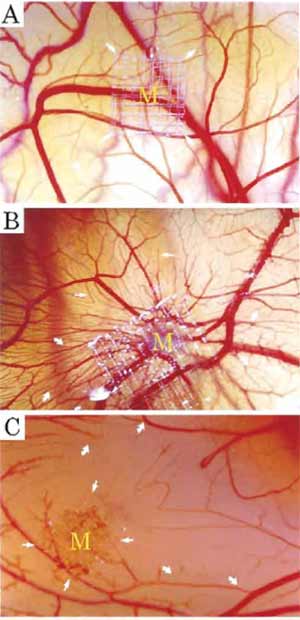 |
| FIGURE 1 (A) Normal chick chorioallantoic membrane after 8- day incubation. (B) A typical example of the CAM stimulated by FGF-2. New vessel sprouting stimulated by 2.5 µg of FGF-2 after a 5-day implantation (arrows). (C) A typical example of inhibition of CAM angiogenesis by 50 µg of epigallocatechin-3-gallate. Formation of avascular zones is marked by arrows. |
- All procedures should be carried out in a 37°C warm room equipped with a sterile ventilation hood and a humidifier.
- Set up the chick incubator at 37°C with 75% humidity.
- Place freshly fertilized eggs onto the selves of the incubator and incubate the eggs for 72h.
- Switch on the humidifier to make sure that the room air has a similar humidity as that in the egg incubator.
- Prewarm plastic tissue dishes (100 × 20mm) at 37°C.
- In the sterile hood, hold the egg in one hand and gently make a small crack using a metal or a glass stick. Open the egg using both thumbs placed on each side of the crack and gently place the contents into a prewarmed tissue culture dish. It is important that the yolk sac remains intact.
- Immediately place the dish into a tissue culture incubator supplied with 3 or 4% CO2 and 75% humidity at 37°C.
- Incubate the embryos for 48h in case of testing inhibitors of angiogenesis. Use 6 days for testing angiogenesis stimulators.
- During the incubation, prepare 0.9% methylcellulose in H2O as autoclaved sterile solution and tested angiogenic or antiangiogenic compounds at appropriate concentrations (e.g., 2mg/ml) with H2O.
- Place a piece of nylon mesh onto a glass beaker in a sterile ventilation hood.
- Mix 10 µl of 0.9% methylcellulose with 10 µl of tested compounds and carefully transfer the mixture onto the nylon mesh (the drop will hang on the mesh).
- When the drop has dried out, carefully cut out the area as a square (about 3 × 3 mm).
- For antiangiogenesis, after a 48-h incubation carefully place the nylon mesh containing the test compounds onto the newly formed chorioallantoic membrane.
- Return the chick embryos to the incubator and incubate the embryos for 1-2 days.
- Detect the antiangiogenic effect of the tested compound under a stereomicroscope.
- For a typical potent angiogenesis inhibitor, the formation of avascular zones around the implant can be detected readily after 24-48h of implantation.
- Testing of angiogenesis stimulators takes place after 6 days of incubation. Carefully place the nylon mesh on the almost fully developed chorioallantoic membrane, preferably at a site with sparse vessel formation.
- Return the chick embryos to the incubator and incubate the embryos for 3-5 days. For a typical potent angiogenic factor, the formation of new microvessel sprouts and branches can now be detected (see Fig. 1).
C. Mouse Corneal Angiogenesis Assay
For additional information, see Cao et al. (2003), Kenyon et al. (1996); and Cao and Cao (1999).
1. Stimulation of Angiogenesis
Steps
- Order male or female mice at the age of between 6 and 9 weeks. For routine angiogenesis analysis, the C57B1/6 strain is commonly used.
- Suspensions of sterile H2O containing the appropriate amount of angiogenic factors such as FGF-2 or VEGF and 10mg of sucralfate.
- Vacuum the mixture solution for 5 min in a speed centrifuge.
- Add 10µl of 12% hydron in ethanol.
- Deposit the suspension onto a nylon mesh (pore size 0.4 × 0.4mm) and embed between the fibers (the total grid area: 15 × 15 mm).
- Cover both sides with a thin layer of hydron and allow the materials to dry at room temperature.
- When the immobilized angiogenic factors are dried on the mesh, pull apart the grid fibers and uniformed pellets (0.4 × 0.4) are released.
- Choose equal sized pellets; each pellet contains equal amounts of angiogenic factors.
- Anesthetize mice with methoxyflurane and anesthetize topically with 0.5% proparacaine.
- Proptose the eye globes with a jeweler's forceps.
- Under an ophthalmological operation microscope, perform a central intrastromal linear keratotomy with a surgical blade.
- Using a modified von Graefe knife (2 × 3mm), dissect a lamellar micropocket toward the temporal limbus.
- The micropocket is usually extended to 1.0-1.2mm of the temporal limbus.
- Place a single pellet on the corneal surface at the base of the pocket with jeweler's forceps.
- Using one arm of the forceps, push the pellet to the end of the pocket.
- Apply erythromycin ointment immediately to the operated eyes.
- Examine the implanted eyes routinely under a slit lamp biomicroscope between days 4 and 7 after pellet implantation.
- Anesthetize mice with methoxyflurane and position the eyes properly.
- Measure the maximal vessel length of neovascularization zone from the limbal vascular plexus toward the pellet with a linear reticule through the microscope (equipped with the microscope).
- Measure the contiguous circumferential zone of neovascularization as clock hours (if the eye is visualized as a clock).
2. Inhibition of Angiogenesis
Steps
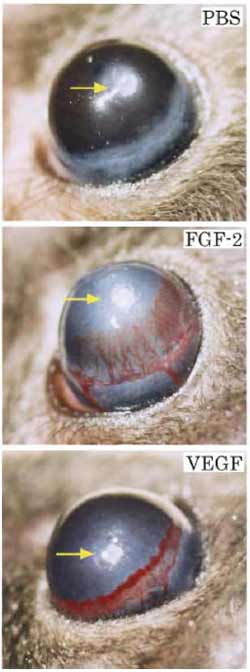 |
| FIGURE 2 Typical examples of FGF-2- and VEGF-induced corneal neovascularization on day 5 after implantation. (Top). PBS buffer and sucralfalte polymer alone without angiogenic factors. (Center) FGF-2 at a 80-ng dose induces intense neovascularization originating from the limbal vessels toward the implanted pellet. (Bottom) VEGF at a 160-ng dose induces a robust neovascularization response. |
- Repeat steps 1-16 described in Section III,C,1.
- Systemically inject potential angiogenesis inhibitors into mice with angiogenic factor-implanted eyes. The injection routes include intravenous, intraperitoneal, subcutaneous, and intramuscular injections. The injected volume usually should not exceed 0.1 ml / 10 g mouse.
- The duration and frequency of treatment are dependent on the potency and the half-lives of tested angiogenesis inhibitors.
- In a control group of mice, inject the relevant buffer that is used to suspend the angiogenesis inhibitors.
- The examination of corneal neovascularization is described in steps 17-20 in Section III,C,1 (see Fig. 2).
IV. COMMENTS
Endothelial cell division is an essential process for the growth of new blood vessels. Therefore, the endothelial cell proliferation assay is the most relevant and reliable assay system used to detect endothelial cell growth. As new blood vessels usually bud from microvessels, endothelial cells isolated from capillaries are most optimal for endothelial cell growth assay. This article described a method that employs BCE cells as primary endothelial cells for the in vitro proliferation assay. Among in vivo angiogenesis models, the mouse corneal angiogenesis model is the most rigorous and clear system to detect angiogenic responses. As the corneal organ is naturally avascular, corneal neovascularization indicates that all blood vessels are newly formed vessels. In contrast to the CAM assay, corneal angiogenesis is devoid of preexisting blood vessels. Thus, this system is convenient for studying new blood vessel formation, new blood vessel stability, and structures of newly formed vessels. For example, a recent study of blood vessel stability in the cornea has found that not all angiogenic factors can stabilize the newly formed vasculature (Cao et al., 2003). As most transgenic or knockout animal models are performed in mice, the mouse corneal model is very valuable in detecting the impact of overproduction or loss of a particular gene on new blood vessel formation. With exceptional skills, the corneal angiogenic responses in knockout mice can be detected in newborns (Zhou et al., 2000). Although the CAM assay is not an ideal quantitative angiogenesis assay, this assay is a fast screening system that allows the determination of angiogenic or antiangiogenic responses within a relatively short time. In addition, the CAM assay requires less sophisticated equipment and less cost.
V. PITFALLS
A. Endothelial Cell Proliferation Assay
- The success of in vitro endothelial cell proliferation assay is entirely dependent on the number of population doublings (PDLs) of the BCE cells. In principle, low PDLs of BCE cells will increase successful rates. Primary BCE cells will stop their responses to angiogenic stimuli around the age of 40 PDLs. Ultimately, these cells enter into a senescent state at the average of 45 PDLs. Therefore, it is important to avoid using presenescent BCE cells in endothelial cell proliferation assay.
- Primary endothelial cells isolated from different tissues or organs may react to different mitogens. Indeed, a tissue-specific endothelial growth factor has been identified (LeCouter et al., 2001). Thus, it is important to determine the stimulatory/inhibitory activity of a compound using primary endothelial cells isolated from different tissues.
- Not all angiogenic factors are able to induce endothelial cell proliferation or migration due to the diverse biological functions of various angiogenic factors. For example, FGF-2 is a potent angiogenic factor that preferentially stimulates endothelial cell proliferation but not cell migration. In contrast, VEGF is a potent endothelial chemotactic factor but a poor endothelial mitogen (Yoshida et al., 1996). Thus, the stimulatory effect of an angiogenic factor should be tested in various endothelial cell assays.
- In endothelial cell inhibition assays, it is critically important that low concentrations of angiogenic factors be used. For example, in the BCE cell assay, usually 1 ng/ml of FGF-2 is used to stimulate cell growth. If higher concentrations of FGF-2 were used, angiogenesis inhibitors would not be able to counteract the stimulatory effect.
B. CAM Assay
- Make sure that the entire experimentation is performed with optimal temperature and humidity. The chick embryos are extremely vulnerable to pathogen infections. Thus, a strict sterile condition is required, including washing hands and wearing gloves when handing eggs and embryos.
- Use fresh fertilized eggs, which should not be more than 3 days old, as the survival rate of embryos will be reduced dramatically.
- Do not shake or crack eggs by hitting them against other solid objects.
- Avoid cutting the embryonic vessels by the sharp edge of the egg shell.
- Do not keep embryos with nonintact yolk.
- Avoid hemorrhages of the CAM during the implantation of meshes.
- The release half-life of small chemical compounds immobilized on the nylon mesh can be different. Thus, it is important to analyze the results at different time points.
C. Mouse Corneal Assay
- Do not choose aged mice that usually produce delayed angiogenic responses.
- Avoid using Balb/C or other white background mice. Their red-color eyes may disturb the quantification of corneal neovascularization.
- Avoid accommodating many mice, especially male mice, in one case. Their unusual behaviors may interrupt corneal neovascularization.
- Prepare corneal implants under sterile conditions, as corneal inflammation could cause neovascularization.
References
Cao, R., Brakenhielm, E., Pawliuk, R., Wariaro, D., Post, M. J., Wahlberg, E., Leboulch, P., and Cao, Y. (2003). Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nature Med. 31, 31.
Cao, R., Brakenhielm, E., Wahlestedt, C., Thyberg, J., and Cao, Y. (2001). Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc. Natl. Acad. Sci. USA 98, 6390-6395.
Cao, R., Wu, H. L., Veitonmaki, N., Linden, P., Farnebo, J., Shi, G. Y., and Cao, Y. (1999). Suppression of angiogenesis and tumor growth by the inhibitor K1-5 generated by plasmin-mediated proteolysis. Proc. Natl. Acad. Sci. USA 96, 5728-5733.
Cao, Y. (2001). Endogenous angiogenesis inhibitors and their therapeutic implications. Int. J. Biochem. Cell. Biol. 33, 357- 369.
Cao, Y., and Cao, R. (1999). Angiogenesis inhibited by drinking tea. Nature 398, 381.
Cao, Y., Chen, A., An, S. S., Ji, R. W., Davidson, D., and Llinas, M. (1997). Kringle 5 of plasminogen is a novel inhibitor of endothelial cell growth. J. Biol. Chem. 272, 22924-22928.
Cao, Y., Ji, R. W., Davidson, D., Schaller, J., Marti, D., Sohndel, S., McCance, S. G., O'Reilly, M. S., Llinas, M., and Folkman, J. (1996). Kringle domains of human angiostatin. Characterization of the anti-proliferative activity on endothelial cells. J. Biol. Chem. 271, 29461-29467.
Cao, Y., Linden, P., Farnebo, J., Cao, R., Eriksson, A., Kumar, V., Qi, J. H., Claesson-Welsh, L., and Alitalo, K. (1998). Vascular endothelial growth factor C induces angiogenesis in vivo. Proc. Natl. Acad. Sci. USA 95, 14389-14394.
Folkman, J. (1995). Angiogenesis in cancer, vascular, rheumatoid and other disease. Nature Med. 1, 27-31.
Hanahan, D., and Folkman, J. (1996). Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353-364.
Jain, R. K., Schlenger, K., Hockel, M., and Yuan, E (1997). Quantitative angiogenesis assays: progress and problems. Nature Med. 3, 1203-1208.
Kenyon, B. M., Voest, E. E., Chen, C. C., Flynn, E., Folkman, J., and D'Amato, R. J. (1996). A model of angiogenesis in the mouse cornea. Invest. Ophthalmol. Vis. Sci. 37, 1625-1632.
LeCouter, J., Kowalski, J., Foster, J., Hass, P., Zhang, Z., Dillard-Telm, L., Frantz, G., Rangell, L., DeGuzman, L., Keller, G. A., Peale, E, Gurney, A., Hillan, K. J., and Ferrara, N. (2001). Identification of an angiogenic mitogen selective for endocrine gland endothelium. Nature 412, 877-884.
Rupnick, M. A., Panigrahy, D., Zhang, C. Y., Dallabrida, S. M., Lowell, B. B., Langer, R., and Folkman, M. J. (2002). Adipose tissue mass can be regulated through the vasculature. Proc. Natl. Acad. Sci. USA 99, 10730-10735.
Yoshida, A., Anand-Apte, B., and Zetter, B. R. (1996). Differential endothelial migration and proliferation to basic fibroblast growth factor and vascular endothelial growth factor. Growth Factors 13, 57-64.
Zhou, Z., Apte, S. S., Soininen, R., Cao, R., Baaklini, G. Y., Rauser, R. W., Wang, J., Cao, Y., and Tryggvason, K. (2000). Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl. Acad. Sci. USA 97, 4052-4057.
Support our developers




