Assays Measuring Membrane Transport in the Endocytic Pathway
I. INTRODUCTIONSignificant progress has been made in understanding mechanisms regulating endocytic membrane traffic using cell-free assays (Braell, 1987; Davey et al., 1985; Diaz et al., 1988; Gruenberg and Howell, 1986; Woodman and Warren, 1988) (see Fig. 1). Both early and late endosomes exhibit homotypic fusion properties in vitro, as in vivo, yet they do not fuse with each other (Aniento et al., 1993). Transport from early to late endosomes is achieved by multivesicular intermediates termed endosomal carrier vesicles (ECV/MVB), which are presumably translocated on microtubules between the two compartments (Aniento et al., 1996; Bomsel et al., 1990; Gruenberg et al., 1989). The vectorial or heterotypic interactions of ECV/MVBs with late endosomes have also been reconstituted in vitro, as has the involvement of microtubules and motor proteins in this process (Aniento et al., 1993; Bomsel et al., 1990). By reducing the components in these in vitro assays to a cytosol source, an ATP-regenerating system, salts, and the purified endosomal membranes, the specificity of endosomal fusion events has been addressed, and the molecules and mechanisms involved have been studied. In fact, a number of conserved molecules, as well as molecules specific for different steps of the endocytic pathway, have been identified and/or characterized using cell-free assays such as those described in this protocol (for review, see Gruenberg and Maxfield, 1995).
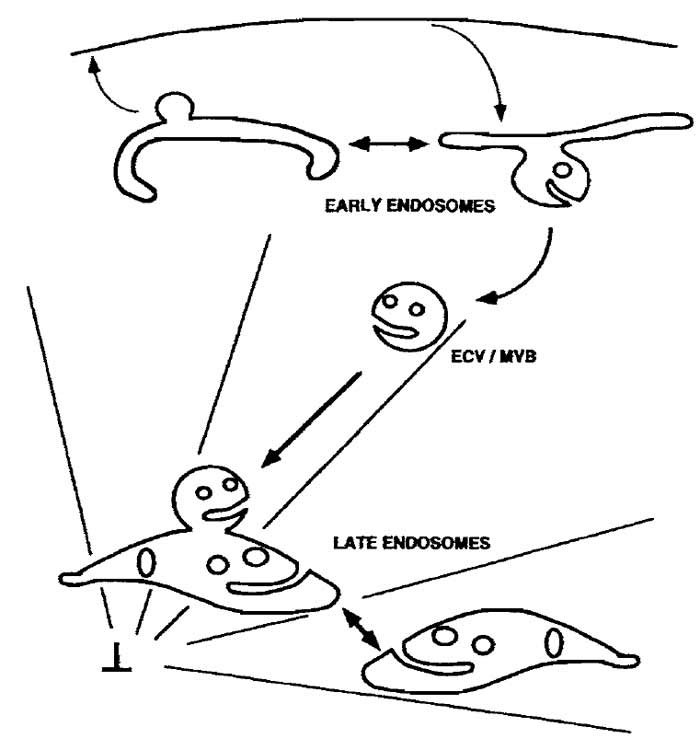 |
| FIGURE 1 Membrane trafficking in the endocytic pathway. The reconstituted steps of the endocytic pathway described in this protocol are the (a) fusion of early endosomes with each other, (b) fusion of ECV/MVBs with late endosomes, and (c) fusion of late endosomes with each other. An in vitro budding assay for the formation of ECV/MVBs from early endosomes, which are competent to fuse with late endosomes, is described in Aniento et al. (1996). As shown, ECV/MVBs are transported along microtubules from early to late endosomes. If microtubules are depolymerized in vivo, prior to the loading of cells with an endocytic tracer, this tracer will accumulate in ECW/MVBs. These vesicles will then fuse with late endosomes, loaded with a different marker, in vitro. |
This assay for endocytic vesicle fusion is based on the formation of a complex resulting from a reaction between two products present in separate populations of endosomes: avidin and biotinylated horseradish peroxidase (bHRP). These reaction products can be internalized into endosomes by fluid phase or receptor-mediated endocytosis in vivo. Avidin and a biotinylated compound are used to provide a fusionspecific reaction because of the high binding affinity and low dissociation constant of avidin for biotin. Following internalization, cells are homogenized and purified endosomal fractions are prepared, which are combined in the assay together with cytosol and ATP. If fusion occurs, a complex is formed between avidin and bHRP. At the end of the assay, the reaction mixture is extracted in detergents in the presence of excess biotinylated insulin as a quenching agent. The avidin-bHRP complex is then detected by immunoprecipitation with antiavidin antibodies, and the enzymatic activity of the bHRP associated with the immunoprecipitate is quantified. This article describes techniques for the preparation and partial purification of three different loaded endosomal fractions from BHK cells: early endosomes, endosomal carrier vesicles, and late endosomes. In addition, this article describes the preparation of the cytosol source used, as well as the techniques for the fusion assays themselves.
II. MATERIAL AND INSTRUMENTATION
Standard laboratory rockers for washing cells and a large 37°C water bath, which can fit a metal plate of dimensions of 20 × 33 cm, are used. Large rectangular ice buckets (Cat. No. 1-6030), from NeoLab GmbH, can also accommodate metal plates of the same dimensions. Cell scrapers (flexible rubber policemen) with a silicone rubber piece of about 2cm, cut at a sharp angle, and attached to a metal bar, are made. A standard low-speed cell centrifuge and Beckman ultracentrifuges and rotors are used. The refractometer (Cat. No. 79729) is from Carl Zeiss Inc., and the pump for collecting sucrose gradients (peristaltic pump P-l) is from Pharmacia Fine Chemicals. A rotating wheel (such as Snijders Model 34528) with a speed of about 10 rotations per minute should be used. All tissue culture reagents, including modified Eagle's medium (MEM), are from either Sigma Chemical Company or GIBCO-BRL/Life Technologies. Peroxidase from horseradish (HRP) (Cat. No. P-8250), ATP (disodium salt, Cat. No. A-5394), and deuterium oxide (D2O, Cat. No. D-4501) are from Sigma Chemical Company, Ltd. Biotinyl-ε-aminocaproic acid N-hydroxysuccinimide ester (biotin-X-NHS, Cat. No. 203188) is from Calbiochem. Avidin (egg white, Cat. No. A-887) is from Molecular Probes. Creatine phosphate (Cat. No. 621714), creatine phosphokinase (Cat. No. 127566), and hexokinase [(NH4)2SO4 precipitate of yeast hexokinase, 1400U/ml, Cat. No. 1426362] are from Boehringer- Mannheim GmbH. Protein A-Sepharose beads (Cat. No. CL-4B) are from Pharmacia. Antiavidin antibodies are generated by injecting purified avidin into rabbits and are affinity purified prior to use. Antiavidin antibodies are also available commercially from several companies. BCA protein assay reagents (Cat. No. 23223) are from Pierce, and Bio-Rad protein assay reagents (Cat. No. 500-0006) are from Bio-Rad Laboratories GmbH.
III. PROCEDURES
A. Internalization of Endocytic Markers into Early Endosomes (EE) from BHK Cells
Solutions
- Internalization media (IM): MEM containing 10 mM HEPES and 5 mM D-glucose, pH 7.4. Filter sterilize and store at 4°C.
- Phosphate-buffered saline (PBS): 137mM NaCI, 2.7 mM KCL, 1.5 mM KH2PO4, and 6.5 mM Na2HPO4; should be pH 7.4. Filter sterilize and store at 4°C.
- Biotinylated horseradish peroxidase: Dissolve 20mg of HRP in 9.5ml of 0.1M NaHCO3/Na2CO3, pH 9.0, buffer (make fresh and check pH carefully) in a small glass Erlenmeyer flask. Dissolve 20mg of biotin-XNHS in 0.5 ml dirnethylformamide. Mix by adding the biotin dropwise to the HRP mixture while gently stirring or shaking the Erlenmeyer and incubate at room temperature with gentle stirring for at least 45min (a 50:1 molar excess of biotin is important). Quench unreacted active groups with 1 ml of 0.2M glycine, pH 8.0 (use KOH to pH), by adding dropwise while mixing, and mix for an additional 15 min at room temperature. Transfer to 4°C. Dialyze the mixture extensively against PBS-or IM at 4°C (at least four changes of 200 ml each time). The final dialysis should be in IM. Measure protein concentration (should be about 2ml/ml) and HRP enzymatic activity (should be unchanged). Aliquot in sterile tubes, freeze in liquid N2, and store at -20°C until use. Immediately before use, thaw quickly and warm to 37°C.
- Avidin: Avidin powder dissolved in IM at 3mg/ ml. Make fresh immediately before use and warm to 37°C.
- PBS/BSA: 5mg/ml BSA in PBS-. Make fresh before use and cool to 4°C.
Steps
- Cell culture: Maintain monolayers of baby hamster kidney (BHK-21) cells as described in Gruenberg et al. (1989). For a fusion assay of 5-10 points, eight petri dishes (10 cm diameter) should be prepared 16 h before the experiment: four for preparing bHRP-labeled EEs and four for preparing avidin-labeled EEs.
- Fluid-phase internalization: Wash each 10-cm dish of cells twice with 5 ml ice-cold PBS-on ice. This and other washes on ice to follow are performed most easily by placing four dishes onto a metal plate in a large ice bucket on a rocker. After the last wash, remove PBS and place the dish on a metal plate in a 37°C water bath. Add at least 3ml/dish bHRP or avidin solution prewarmed to 37°C. Incubate for 5 min.
- Washes: From now on, all work should be done at 4°C or on ice. Return the dishes to the metal plate in the ice bucket. Remove the avidin or bHRP solution and wash dishes three times for 5 min with 5 ml icecold PBS/BSA followed by 2 × 5 min with 5 ml ice-cold PBS.
- Homogenization and fractionation: Go directly to Section IIIC.
B. Internalization of Endocytic Markers into Endosomal Carrier Vesicles (ECV) and Late Endosomes (LE)
Solutions
- Nocodazole stock: 10mM in dimethyl sulfoxide (DMSO), aliquoted, and stored at -20°C.
- IM/BSA: IM containing 2mg/ml BSA. Make fresh before use and warm to 37°C.
- All solutions listed in Section III,A.
Steps
- Cell culture: For a fusion assay of 5-10 points, 10 dishes (10cm) of BHK cells should be prepared as described in Section III,A, step 1. For ECV-LE fusion assays, use 5 dishes for bHRP-labeled ECVs and 5 dishes for avidin-labeled LEs, For LE-LE fusion assays, use 5 dishes for bHRP-labeled LEs and 5 dishes for avidin-labeled LEs.
- Nocodazole pretreatment for ECV preparation: Intact microtubules are required for the delivery of endocytosed markers to the LE. Therefore, markers accumulate in transport intermediates (ECVs) in the absence of microtubules. Whereas stable microtubules are cold sensitive, dynamic microtubules are depolymerized easily in the presence of nocodazole (Aniento et al., 1993; Bomsel et al., 1990). In BHK cells, microtubules can be depolymerized efficiently in the presence of nocodazole, whereas cold treatment is without effect. For ECV preparation, depolymerize the microtubules immediately before the experiment with 10µM nocodazole at 37°C for 1-2 h in media used to grow cells in a 5% CO2 incubator. Following this step, nocodazole (10µM) should remain present in all solutions up to the homogenization step. For LE preparation, do not treat with nocodazole or include nocodazole in any solutions.
- Fluid-phase internalization: Wash each 10-cm dish of cells twice with 5 ml ice-cold PBS+/- 10µM nocodazole on ice, as in Section III,A, step 2. After the last wash, remove the PBS and place the dish on a metal plate in a 37°C water bath. Add at least 3 ml bHRP or avidin solution for making LEs or bHRP + 10 µM nocodazole for making ECVs. Incubate for 10min.
- Chase: Remove bHRP or avidin and wash twice quickly at 37°C with 10ml PBS/BSA+/- 10µM nocodazole, prewarmed to 37°C. Remove last wash, and add 8ml IM/BSA+/- 10µM nocodazole, prewarmed to 37°C. Incubate at 37°C (in water bath or in a 37°C incubator without CO2) for 45 min.
- Washes: Remove IM/BSA, move dishes to ice bucket, and wash 2 × 5min with 5ml cold PBS/BSA followed by 5 min with 5 ml cold PBS on ice.
C. Homogenization and Fractionation of Cells
Solutions
- PBS: See Section IIIA.
- 300mM imidazole stock: Dissolve imidazole in H2O and adjust pH to 7.4 with NaOH, filter sterilize, and store at 4°C
- Homogenization buffer (HB): Add imidazole from 300mM stock to H2O and dissolve sucrose such that the final concentrations are 250 mM sucrose and 3 mM imidazole. Filter sterilize and store at 4°C.
- 62% sucrose solution: For 100ml, add 1 ml of imidazole from 300mM stock to 15ml H2O. Add 80.4g sucrose and dissolve by stirring at 37°C. Add H2O and mix until the refractive index is 1.4464.
- 10 and 16% sucrose solutions in D2O: For 100 ml, add 1 ml imidazole from 300mM stock to 50ml D2O. For 10% solution, add 10.4g sucrose, and for 16% solution, add 17.0g sucrose. Dissolve sucrose, add D2O, and mix until the refractive index is 1.3479 for the 10% solution and 1.3573 for the 16% solution.
Steps
- Cell scraping: All of the following steps should be performed on ice or at 4°C. After the last wash, remove all PBS. Add 2ml/dish PBS and rock the dish so that cells do not dry. Using a flexible rubber policeman, scrape round 10-cm dishes by first scraping in a circular motion around the outside of the dish, followed by a downward motion in the middle of the dish. Scrape gently in order to obtain "sheets" of cells. Using a plastic Pasteur pipette, gently transfer the scraped "sheets" of cells from four or five dishes into a 15-ml tube on ice.
- Centrifuge at 1200rpm for 5min at 4°C. Gently remove supernatant.
- Add 1 ml HB to pellet, using a plastic Pasteur pipette, gently pipette up and down one time and add an excess of HB (4-5ml) to change buffer. Centrifuge again at 2500rpm for 10min at 4°C. Remove supernatant.
- Homogenization: It is important that cells are homogenized under conditions where endosomes are released from cells, yet where latency is high so that the endosomes are not broken and retain their internalized marker. First add 0.5 ml HB to the cell pellet. Using a 1-ml pipetman, gently pipette up and down until the pellet is resuspended and particles can no longer be seen by eye. Do not introduce air bubbles. Using a 22-gauge needle connected to a narrow 1-ml Tubercutine syringe, prewet the needle and syringe with HB so that no air is introduced. Insert the needle into the cell homogenate, slowly pull up on the syringe until most of the cell homogenate is in the syringe, and gently expel without bubbles. Repeat this procedure until plasma membranes are broken, yet nuclear membranes are not. Monitor homogenization as follows. Take 3µl of homogenate and place in a 50-µl drop of HB on a glass slide. Mix and cover with a glass coverslip. Observe by phase-contrast microscopy, using a 20× objective. Homogenize until unbroken cells are no longer observed, yet nuclei, which appear as dark round or oblong structures, are not broken. Usually between 3 and 10 up-and-down strokes through the needle are necessary. Centrifuge homogenate at 2000rpm for 10min at 4°C and carefully collect the postnuclear supernatant (PNS) and nuclear pellet.
- Save a 50-µl aliquot of each PNS fraction for measuring latency and for calculating the balance sheet as described in Section III,D. Adjust the sucrose concentration of the remaining PNS to 40.6% by adding about 1.1 volume of 62% sucrose solution per volume of PNS. Mix gently but thoroughly, without bubbles. Check sucrose concentration using a refractometer.
- Place adjusted PNS in the bottom of a SW60 centrifuge tube. On top of the PNS, layer 1.5 ml of 16% sucrose solution in D2O, followed by 1 ml of 10% sucrose solution in D2O, and fill tube with HB. Steps should be layered so that interfaces are clearly seen and not disturbed. See Gruenberg and Gorvel (1992) for diagram of gradients.
- Centrifuge gradients in SW60 rotor at 35,000rpm for 1 h at 4°C.
- Carefully remove the interfaces from the gradients after centrifugation by first placing gradients in a test tube rack with a black backdrop. The interfaces should appear white. The layer of white lipids on top of the gradient should be removed carefully. Collect fractions at 4°C using a peristaltic pump at speed 2, with capillary tubes connected to each end. Place the outgoing end into a collection tube and collect the top interface carefully (10%/HB interface = LE + ECV fraction) first. Collect by holding the capillary tube directly in the middle of the wide interface and slowly move in a circular motion until most of the white interface is collected into the smallest possible volume. Wash the pump tubing with water and then collect the EE (16/10%) interface into another tube. Fractions can be frozen and stored in liquid N2 until use in fusion assays if they are carefully frozen quickly in liquid N2 and thawed quickly at 37°C immediately before use.
D. Measurement of Latency and Balance Sheet for Gradients
Solutions
- HRP stocks: 1-10ng HRP in 0.1ml HB, for standards.
- HB: See Section IIIC.
- HRP reagent: 0.342mM o-dianisidine and 0.003% H2O2 in 0.05M Na-phosphate buffer, pH 5.0, containing 0.3% Triton X-100. To prepare, use very clean glassware or plasticware (as in for tissue culture) and mix 12 ml of 0.5M Na-phosphate buffer, pH 5.0 (filter sterilized), and 6ml of 2% Triton X-100 (filter sterilized) with 111 ml sterile H2O. Add 13mg o-dianisidine, dissolve gently, and add 1.2ml 0.3% H2O2 (filter sterilized). Avoid magnetic stirring. Solution should be clear. Store at 4°C in the dark.
- 1 mM KCN in H2O
- Protein assay system (such as the BCA protein assay reagent or the Bio-Rad protein assay system)
Steps
- Load a 20-µl aliquot of bHRP PNS into an airfuge tube or a small tabletop ultracentrifuge tube of the Beckman TL-100 type and fill the tube with a known volme of HB. Mix thoroughly by pipetting without air bubbles. Centrifuge at 4°C for 20min at 20psi in an airfuge or at 200,000g for 20min in a tabletop ultracentrifuge rotor (such as Beckman TLA-100.1). Transfer the supernatant to another tube. Resuspend the pellet in 50 µl HB.
- To measure the latency, adjust samples, blanks, and standards with HB so that the final volume of each is 0.1 ml. Assay both the pellet and the supernatant of the latency measurement. If the supernatant volume is over 0.1ml, assay only 0.1ml. Add 0.9ml of HRP reagent to each tube, mix quickly, and record the time with a stop clock. Allow color to develop in the dark, as this reagent is light sensitive. When a brown color has begun to develop, read the absorbance at 455 nm and record the time (results expressed as OD units/ min or ng HRP/min). Stop the reaction with 10µl of 1.0 mM KCN if necessary.
- Calculate latency by first adding the value (OD/min) for HRP in the pellet to that of HRP in the supernatant (OD/min after correcting for total supernatant volume). The value for the pellet divided by the total value is the percentage latency. Latency should be over 70% in order to measure endosome fusion.
- The amount of HRP in each gradient fraction collected from the bHRP gradient can be measured by assaying an aliquot (about 50µl) of each fraction as described in step 2.
- Measure the amount of protein in each gradient fraction using a standard protein assay system, as described in the manual.
- Calculate percentage yield (percentage of HRP in each fraction compared to total amount of HRP in PNS), specific activity (SA) (HRP activity per unit protein), and relative specific activity (RSA) (divide specific activity of each fraction by the specific activity of the PNS). See Gruenberg and Gorvel (1992) for an example of a typical balance sheet.
E. Preparation of BHK Cell Cytosol
Solutions
- PBS-: See Section IliA.
- HB: See Section IIIC.
- HB + protease inhibitors: HB with the following protease inhibitors added immediately before use: 10µM leupeptin, 1 µM pepstatin A, 10ng/ml aprotinin, and, if needed, 1 µm phenylmethylsulfonyl fluoride.
Steps
Two possible cytosol sources for all of the assays described are BHK and rat liver cytosol. For rat liver cytosol preparation, refer to Aniento et al. (1993).
- BHK cells, maintained as described in Section IIIA, should be plated approximately 16h before the experiment. Large (245 x 245 x 25 mm) square dishes are convenient for large cytosol preparations.
- All steps should be performed on ice or at 4°C. Wash dishes four times with excess PBS (50 ml per dish for large square dishes).
- Remove PBS from the last wash, add 12ml PBS per dish, and rock the dish so that cells do not dry. Scrape cells with a rubber policeman using firm, downward motions, going from top to bottom while holding the plate at an angle, as described in Section IIIC, step 1.
- Collect scraped cells into 15-ml tubes (one tube per dish). Centrifuge at 1200rpm for 5 min at 4°C.
- Remove supernatant and gently add 5ml HB with a plastic Pasteur pipette and pipette up and down one time.
- Centrifuge at 2500 rpm for 10 min at 4°C. Remove supernatant and resuspend pellet in 1.2ml HB + protease inhibitors. Separate into two tubes (about 0.7ml/tube) for homogenization and homogenize as described in step 4 of Section IIIC.
- Centrifuge at 2500rpm for 15min at 4°C. Add supernatant (PNS) to a centrifuge tube for the TLS-55 rotor (for the Beckman TL-100 tabletop ultracentrifuge) and centrifuge in TLS-55 for 45 min at 55,000 rpm at 4°C. Remove fat from the top using an aspirator. Transfer supernatant (cytosol fraction) to a new tube without disturbing the pellet. Determine the protein concentration of supernatant. Cytosol should be at least 15 mg/ml to give a good signal for fusion assays. Aliquot on ice, freeze quickly, and store in liquid N2 until use.
F. Preparation of Antiavidin Beads for the in Vitro Fusion Assay Described in Section IIIG
Solutions
- PBS/BSA: Dissolve 5 mg/ml BSA in PBS. Filter sterilize and store at 4°C.
- Sterile PBS: PBS as described in Section III,A, filter sterilize or autoclave, and store at 4°C.
- Antiavidin antibody: Affinity purify and store aliquoted in 50% glycerol/PBS at -20°C.
Steps
To determine how many antiavidin beads to prepare, first determine the number of fusion assay points. From a typical gradient (see Gruenberg and Gorvel, 1992) about 150µg of EE and 70µg of ECV or LE are obtained. Optimal amounts of endosomes to use for fusion assays are 20µg of each EE fraction and 10µg of each ECV or LE fraction. Therefore, a typical experiment (one gradient each of avidin and bHRPlabeled fractions) will provide enough endosomes for about seven fusion assay points.
- Swell 1.5 g of protein A-Sepharose beads in 10 ml sterile H2O at room temperature overnight.
- Wash beads three times in 10ml sterile PBS by centrifuging beads in 15-ml tubes at 3000rpm for 2 min, resuspending in PBS each time.
- After the final wash, resuspend beads in an equal volume of sterile PBS per volume of packed beads. Store beads this way up to several months at 4°C.
- One hundred microliters of this 1:1 slurry is required per fusion assay point. Therefore, for 10 assay points, block 1 ml of beads by washing 3× in 10ml PBS/BSA, as described in step 2.
- After final wash, resuspend beads in 10 ml PBS/ BSA. For 10 assay points, add 50 µg of antiavidin antibody (5µg per 100-µl beads). Rotate tube for at least 5 h at 4°C.
- Wash beads four times in PBS/BSA. After the last wash, for 10 assay points, resuspend beads in 10ml PBS/BSA.
- Aliquot 1 ml to each of 10 labeled Eppendorf tubes. Centrifuge in Eppendorf centrifuge at maximum speed for 2 min. Remove supernatant. Beads are now ready for the immunoprecipitation step of the fusion assay (Section III,G, step 10).
G. in vitro Assay of Endocytic Vesicle Fusion
Solutions
- 50× salts: 0.625M HEPES, 75mM Mg-acetate, 50mM dithiothreitol, pH 7, with KOH. Filter sterilize, aliquot, and store at -20°C.
- K-acetate (KOAc stock): 1M in H2O. Filter sterilize, aliquot, and store at -20°C. Note: Depending on the counterion requirement of the experiment, KOAc must be replaced by KCl (see Aniento et al., 1993).
- Biotinylated insulin: 1 mg/ml in H2O. Store at 4°C.
- AT?-regenerating system (ATP-RS): Mix 1:1:1
volumes of the following immediately before use.
- 100 mM ATT: Dissolve in ice-cold H2O, titrate to pH 7.0 with 1M NaOH, filter sterilize, aliquot on ice, and store at -20°C.
- 800 mM creatine phosphate: Dissolve in ice-cold H2O, filter sterilize, aliquot on ice, and store at -20°C.
- 4 mg/ml creatine phosphokinase: To make 4 ml, add 80 p\ of 0.5M NaHPO4 buffer, pH 7.0, to 1.6 ml H2O on ice. When cool, add 16 mg creatine phosphokinase. Vortex until dissolved. Add 2.3 ml ice-cold 87% glycerol. Vortex until well mixed. Aliquot on ice and store at -20°C.
- Hexokinase: Vortex the suspension, pipette the desired amount (e.g., 10µg-0.1mg for one assay point), centrifuge for 2 min in Eppendorf at maximum speed, and aspirate supernatant. Dissolve pellet in the same volume of 0.25M D-glucose. Prepare immediately before use.
- TxlOO stock: 10% stock of Triton X-100 in H2O
- PBS/BSA and sterile PBS: See Section IIIE
- HEP reagent: See Section IIID.
- PBS/BSA/TxlOO: PBS/BSA containing 0.2% Triton X-100, make immediately before use
Steps
- For each fusion assay, at least three points should be included:mATP, +ATP, and the total. To determine fusion efficiency, determine the total (maximal possible fusion value) by mixing 50 //I of each endosomal fraction in an Eppendorf tube on ice. Add 25 µl Txl00 stock and vortex well. Leave on ice at least 30min and add PBS/BSA and continue as described in step 9.
- For all other fusion assay points, 3 µl of 50× salts, 8 µl of biotinylated insulin, and 11 µl of KOAc stock are needed for each point. Make a mixture of these three components by multiplying the number of assay points by 3, 8, and 11 and mix the respective amounts of each component together in one tube. Number Eppendorf tubes for the appropriate number of assay points and put them on ice. Add 22µl of the aforementioned mixture to each tube.
- Add 50µl (750µg-1 mg) of cytosol to each tube and mix.
- Add either 5 µl of ATP-RS or 10 µl of hexokinase to each tube, as appropriate.
- Add 50 µl (7-25 µg) of bHRP-labeled endosomes and 50µl (7-25µg) of avidin-labeled endosomes to each tube. Endosomal fractions from the gradients can be diluted in HB prior to this step, if desired. Mix gently; avoid introducing air bubbles. Leave tubes on ice for 3 min.
- Transfer tubes to 37°C for 45 min. Avoid agitation during this time.
- Return tubes to ice. Add 5µl of biotinylated insulin to each tube and mix.
- Add 25 µl of Txl00 stock and vortex well. Leave tubes on ice for 30min.
- Add 1 ml of sterile PBS/BSA to each tube and mix well.
- Centrifuge for 2min at maximum speed in an Eppendorf centrifuge. Transfer supernatants to numbered tubes containing antiavidin beads, prepared as in Section IIIE
- Rotate beads for at least 5 h at 4°C.
- Centrifuge in Eppendorf centrifuge at maximum speed for 2min. Remove supernatant and wash four times with PBS/BSA/Txl00. Wash once with sterile PBS.
- Remove final supernatant and add 900 //I of HRP reagent to each tube. Allow color to develop in the dark at room temperature. Vortex periodically for 2-3 h or put tubes on rotating wheel in the dark at room temperature while color develops.
- Centrifuge tubes for 2min in Eppendorf centrifuge. Measure the absorbance of the supernatants at 455 nm.
IV. COMMENTS
Refer to Gruenberg and Gorvel (1992) for an example of a typical balance sheet for the sucrose gradient fractionation step. Typical results for fusion assays are shown in Fig. 2.
Highly purified loaded endosomes can be prepared by immunoisolation as described in Howell et al. (1989). Immunoisolated endosomes can then be used in the fusion assays described in Section IIIG. See Gruenberg and Gorvel (1992) and Howell et al. (1989) for details.
ECV-LE fusion is stimulated by the addition of polymerized microtubules to the fusion assay. Endogenous microtubules can be polymerized by adding 20µM taxol to the fusion assay. The preparation of microtubules is described in the article by Ashford and Hyman. See Fig. 2 and Aniento et al. (1993) for more details on the effects of microtubules and MAPs on ECV-LE fusion.
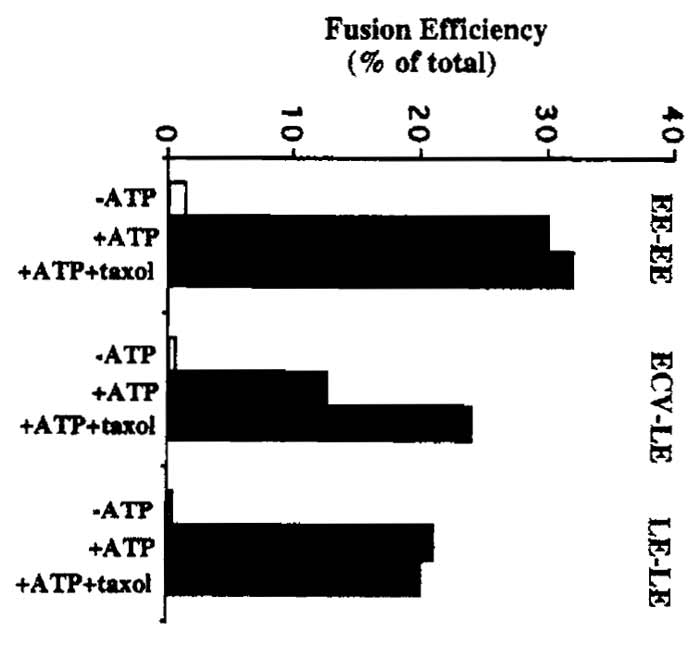 |
| FIGURE 2 Typical fusion assay results. Fusion efficiency is
expressed as a percentage of total fusion between each set of endosomal
membranes. Total, or maximal, endosome fusion is measured
by mixing bHRP and avidin-containing endosomal fractions together
in the presence of detergent, followed by immunoprecipitation
with antiavidin antibodies and HRP determination. Typical
"total" values (measured as absorbance at 455 nm) are in the range
of 0.6-1.0 ∧455 units for EE fusion assays and 0.3-0.7 ∧455 units for
ECV and LE fusion assays. As a control for nonspecific reactions, the
assay is typically carried out without ATP (-ATP). As shown, the
polymerization of endogenous tubulin present in the cytosol in
the presence of taxol is sufficient to facilitate interactions between ECVs and LEs (see Aniento et al., 1993). |
V. PITFALLS
For ECV and LE preparations, cells should be homogenized until vesicles are no longer seen around the periphery of nuclei. If nuclei begin to aggregate during homogenization, however, this is a sign that some are broken as free DNA causes aggregation. Freezing and thawing of endosomes may cause a partial loss in latency. Use very clean plasticware or glassware for all fusion assay manipulations as HRP contamination can occur easily. Nocodazole, o-dianisidine, and KCN are very toxic.
References
Aniento, E, Emans, N., Griffiths, G., and Gruenberg, J. (1993). Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol. 123, 1373-1387.
Aniento, E, Gu, E, Parton, R. G., and Gruenberg, J. (1996). An endosomal /3COP is implicated in the pH-dependent formation of transport vesicles destined for late endosomes. J. Cell Biol. 133, 29-41.
Aniento, E, Roche, E., Cuervo, A., and Knecht, E. (1993). Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Biol Chem. 268, 10463-10470.
Bomsel, M., Parton, R., Kuznetsov, S. A., Schroer, T. A., and Gruenberg J. (1990). Microtubule- and motor-dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell 62, 719-731.
Braell, W. A. (1987). Fusion between endocytic vesicles in a cell-free system. Proc. Natl. Acad. Sci. USA 84, 1137-1141.
Davey, J. S., Hurtley, S. M., and Warren, G. (1985). Reconstitution of an endocytic fusion event in a cell-free system. Cell 43, 643-652.
Diaz, R., Mayorga, L., and Stahl, P. D. (1988). in vitro fusion of endosomes following receptor-mediated endocytosis. J. Biol. Chem. 263, 6093-6100.
Gruenberg, J., and Gorvel, J.-P. (1992). in vitro reconstitution of endocytic vesicle fusion. In "Protein Targetting, a Practical Approach" (A. I. Magee and T. Wileman, eds.), pp. 187-216. University Press, Oxford.
Gruenberg, J., Griffiths, G., and Howell, K. E. (1989). Characterization of the early endosome and putative endocytic carrier vesicles in vivo with an assay of vesicle fusion in vitro. J. Cell Biol. 108, 1301-1316.
Gruenberg, J., and Howell, K. E. (1986). Reconstitution of vesicle fusions occurring in endocytosis with a cell-free system. EMBO J. 5, 3091-3101.
Gruenberg, J., and Maxfield, E R. (1995). Membrane transport in the endocytic pathway. Curr. Opin. Cell Biol. 7, 552-563.
Howell, K. E., Schmid, R., Ugelstad, J., and Gruenberg J. (1989). Immuno-isolation using magnetic solid supports: Subcellular fractionation for cell-free functional studies. Methods Cell Biol 31A, 264-292.
Woodman, P. G., and Warren, G. (1988). Fusion between vesicles from the pathway of receptor-mediated endocytosis in a cell-free system. Eur. J. Biochem. 173, 101-108.
Support our developers




