Cultivation and Retroviral Infection of Human Epidermal Keratinocytes
I. INTRODUCTIONThere are many techniques for culturing human epidermal keratinocytes, but the method described here is the one devised by Rheinwald and Green (1975). With this method, keratinocytes grow as multilayered sheets in which proliferation is confined to the basal layer and terminal differentiation takes place in the suprabasal layers, thus mimicking the spatial organization of normal interfollicular epidermis. These cultures have a range of applications in basic research and in the clinic. They are used to study the factors that regulate stem cell proliferation, terminal differentiation, and tissue assembly, as well as the events that take place during neoplastic transformation (Jones and Watt, 1993; Zhu and Watt, 1996; Levy et al., 2000; Dajee et al., 2003). Practical applications include the treatment of burns victims with cultured autografts (Compton et al., 1989; Ronfard et al., 2000) and the use of transduced keratinocytes as vehicles for gene therapy (Gerrard et al., 1993; Dellambra et al., 2001; Ortiz-Urda et al., 2002).
What follows is a description of the procedures our laboratory uses to initiate and maintain cultures of keratinocytes from neonatal foreskins; it is based on the original Rheinwald and Green method, improved over the years as described by Rheinwald (1989). The key component of the culture system is the presence of a feeder layer of 3T3 cells that supports the growth of keratinocytes from clonal seeding densities (see Fig. 1). In the laboratory, our preferred method for manipulating gene expression in keratinocytes is by retroviral infection (Zhu and Watt, 1996; Levy et al., 1998; Zhu and Watt, 1999) and we have therefore included our current protocol.
II. MATERIALS AND INSTRUMENTATION
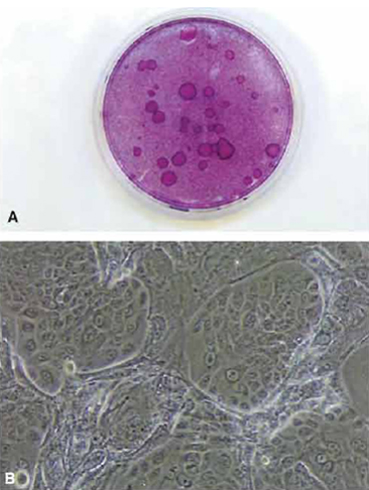 |
| FIGURE 1 (A) Dish of cultured human keratinocytes plated on a feeder layer of 3T3 J2 cells and stained with rhodanile B. Individual clones of keratinocytes are seen as large round plaques. (B) Phasecontrast view of clones of cultured human keratinocytes on a feeder layer of 3T3 J2 cells. |
III. PROCEDURES
A. Feeder Layer
1. Solutions
- Culture medium for 3T3 J2 cells: This consists of DMEM supplemented with 100 IU/ml penicillin, 100gg/ml streptomycin, and 10% bovine serum. It is essential to batch test the serum for optimal growth of 3T3 J2.
- Trypsin-EDTA: Mix one part 0.25% trypsin and four parts 0.02% EDTA. The same solution is used to harvest keratinocytes.
- Phosphate-buffered saline: To make 1 litre, dissolve 0.2g KCl, 0.2g KH2PO4, 8.0g NaCl, and 2.16g Na2HPO4.7H2O in 900ml distilled water. Adjust pH to 7.4, add distilled water to 1-1itre final volume, autoclave, and store at room temperature.
- Mitomycin C in PBS: Prepare a stock solution of 0.4mg/ml in PBS. Filter sterilize and store in aliquots at -20°C.
1. Cells
The J2 clone of random-bred Swiss mouse 3T3 cells was selected to provide optimal feeder support of keratinocytes (Rheinwald, 1989). The cells are maintained by weekly passaging at 1:10 to 1:20 dilution. Fresh cells are thawed every 3 months, as with prolonged passaging the cells start to senesce or undergo spontaneous transformation.
2. Preparing the Feeder Layer
Steps
- To irreversibly inhibit proliferation, add mitomycin C (final concentration, 4µg/ml) to confluent flasks of 3T3 J2 and incubate for 2h at 37°C.
- Remove the medium, rinse the cells once with 0.02% EDTA, and then harvest in trypsin-EDTA. The optimal density of the feeder layer is one-third confluent (Rheinwald and Green, 1975; Rheinwald, 1989); hence each mitomycin C-treated flask is split 1:3.
- The feeders can be used immediately (i.e., plated at the same time as the keratinocytes) or prepared 1-2 days before they are required; if prepared in advance, maintain in DMEM + 10% BS. The feeder layer should not be prepared more than 2 days before use because the feeder cells will start to degenerate.
B. Keratinocyte Culture Medium
Solutions
- Hydrocortisone: Prepare a 5 mg/ml stock in absolute ethanol. Store at-20°C.
- Cholera enterotoxin: Prepare a 10-5M stock in distilled water. Store at 4°C.
- EGF: Prepare at 100gg/ml stock in FAD + FCS . Store at-20°C.
- Insulin: Prepare a 5mg/ml solution in 5mM hydrochloric acid. Store at-20°C.
Steps
- The basic medium consists of three parts DMEM and one part F12 supplemented with 1.8 × 10-4M adenine (FAD), 100IU/ml penicillin, and 100µg/ml streptomycin. Store at 4°C.
- Supplement FAD with 10% FCS. It is essential to batch test the serum for its ability to support high colony-forming efficiency, rapid growth, and serial passage of keratinocytes. Serum batches optimal for keratinocytes tend to be completely unsuitable for fibroblastic cells or hybridomas. Store FCS at-20°C before use.
- Supplement FAD + FCS further with a HICE cocktail consisting of hydrocortisone (0.5µg/ml), insulin (5µg/ml), cholera enterotoxin (10-10M), and EGF (10ng/ml) (all final concentrations).
- Store complete medium (FAD + FCS + HICE) at 4°C and use within 1 week.
C. Source of Keratinocytes
The usual source of keratinocytes is neonatal foreskin obtained, with ethical approval, from routine circumcisions. When handling any human tissue it is essential to take appropriate precautions against the transmission of infectious agents. Obtain the foreskin as soon as possible after circumcision and transfer to the laboratory dry in a sterile Bijou. If it cannot be used immediately, store overnight at 4°C.
D. Isolation of Keratinocytes
Solutions
1. PBS containing 100 IU/ml penicillin and 100µg/ml streptomycin. Other solutions are as described in Sections III,A and III,B.
Steps
- Rinse the foreskin thoroughly in PBS containing 100 IU/ml penicillin and 100µg/ml streptomycin.
- Transfer the tissue to a 100-mm-diameter petri dish, epidermis down. Remove as much connective tissue (muscle and dermis) as possible using sterile curved scissors.
- Transfer the epidermis and remaining connective tissue to a fresh dish and chop into fine pieces (1-3 mm2) using scalpels.
- Flood the dish with 10 ml trypsin-EDTA and transfer the solution containing pieces of skin with a wide-bore pipette to a sterile CelStir. A CelStir is an autoclavable glass vessel containing a magnet suspended by a rod from the lid. Solutions are introduced and removed via a side arm in the vessel.
- Incubate the CelStir at 37°C for 30min on a magnetic stirrer; allow the lumps of tissue to settle out and remove the supernatant.
- Add fresh trypsin-EDTA and repeat the procedure.
- Determine the number of cells in the supernatant with a haemocytometer. Collect the cells by centrifugation and resuspend in FAD + FCS + HICE. There are usually no cells from the first incubation and then 1-5 × 106 cells per subsequent incubation.
- After four or five incubations the cell yield starts to decline; discard any remaining lumps of tissue.
- Pool cells from each incubation and seed onto the feeder layer at a density of 5 × 105 per 75-cm2 flask. The average yield per foreskin is approximately 5 × 106- 107 cells.
- After 2-3 days, small colonies of keratinocytes are visible, surrounded by 3T3 J2 feeder cells. Over the following weeks, individual colonies expand, displacing the feeder layer and merging with one another. At confluence, virtually no feeder cells remain. Feed the cells with fresh medium three times per week.
E. Passaging Keratinocytes
Solutions are as described in Sections III,A and III, B.
Steps
- Passage keratin0cytes prior to confluence (approximately 7-10 days after plating). Remove the medium and rinse the cultures once with 0.02% EDTA.
- Add fresh EDTA and incubate the cultures at 37°C for about 5 min. Then selectively detach the feeders by gentle aspiration with a pipette (Rheinwald, 1989).
- To detach the keratinocytes, add tryspin-EDTA (see Section III,A, solution 2) to the flasks and incubate at 37°C for about 10 min.
- Transfer the cells to a centrifuge tube; use a small volume of culture medium to rinse the flask and then add it to the tube, inactivating the trypsin.
- Recover the cells by centrifugation, count in a haemocytometer, and resuspend in complete medium.
- Cells can be passaged at a density of 1-2 × 105 per 75-cm2 flask containing 3T3 J2 feeders.
F. Frozen Stocks of Keratinocytes and 3T3 J2 Cells
Solution
1. 10% DMSO/90% fetal calf serum
Steps
- Most of the primary keratinocyte cultures are used to prepare frozen stocks; most experiments are carried out on second and subsequent passages.
- For freezing, harvest the keratinocytes as described earlier, but resuspend at 106 / ml in 10% DMSO / 90% foetal calf serum.
- Place 1 ml of cell suspension in each 1.8-ml CryoTube.
- Place the tubes in a rubber rack wrapped in cotton wool overnight at-70°C and then transfer to liquid nitrogen
- Freeze the 3T3 J2 cells in the same way.
G. Preparation of Stable Packaging Lines for Retroviral Vectors
Solutions
- Culture medium for packaging lines: This consists of DMEM supplemented with 100 IU/ml penicillin, 100 µg/ml streptomycin, and 10% heat-inactivated foetal calf serum. The serum is heat inactivated in a water bath at 56°C. for 45 min and can then be stored frozen in aliquots. Heat inactivation of FCS improves retroviral titres.
- Transfection solutions: 50mM chloroquine (2000x) in dH20, 2M CaCl2 in dH20, and 2xHBS, pH 7.0. Filter sterilise each solution, aliquot, and store at-20°C.
- 2xHBS: This is made up with 8.0g NaCl, 6.5g HEPES, and 10ml Na2HPO4 stock solution (5.25g in 500 ml of water). Adjust the pH to 7.0 using NaOH or HCl, bring the volume up to 500 ml, and check the pH again.
- Polybrene: Prepare a 5mg/ml stock solution in PBS. Filter sterilise, aliquot, and store at-20°C.
- Puromycin: Prepare a 2mg/ml stock solution in PBS. Filter sterilise, aliquot, and store at-20°C.
Cells
Phoenix E (ecotropic) packaging cells (Swift et al., 1999) are obtained from ATCC with prior approval of G. Nolan (Stanford University, USA). The AM12 amphotropic packaging line was generated by Markowitz et al. (1988). Packaging cells are maintained by passage at 1:5 (Phoenix E) or 1:10 (AM12) dilution. For optimal virus production, Phoenix cells should initially be reselected sequentially for 4 weeks, 2 weeks in 1 µg/ml diphtheria toxin, and then two weeks in 500 µg/ml hygromycin to increase Envelope and Gag-Pol expression (http: //www.stanford.edu/group/nolan/ index.html). Packaging cells are frozen at 106/ml in 10% DMSO/90% foetal calf serum as described (in Section III, F,) and fresh cells are thawed every 3 months. J2 puro are 3T3 J2 cells that have been transfected with pBabe puro to render them resistant to puromycin (Levy et al., 1998).
Generation of Stably Transduced AM12 Cells
Packaging lines that are generated by retroviral infection tend to have higher viral titres than those generated by transfection (Levy et al., 1998). Therefore, we use a two-step procedure in which Phoenix E cells are transiently transfected with retroviral vector, and packaged virus released by Phoenix E cells is used to infect AM12 cells.
Steps
- Twenty-four hours prior to transfection, plate 5 × 106 Phoenix E cells into one 10-cm-diameter petri dish in 10 ml medium.
- Add chloroquine (final concentration 25µM) to each plate 5 min before transfection. Add 20µg retroviral vector DNA (we routinely use pBabe puro; Morgenstern and Land, 1990; Levy et al., 1998) to 61 µl 2M CaCl2 and make up to a 500µl total volume with ddH20. Add 500µl 2×HBS quickly and then bubble vigorously with an automatic pipettor for 15s. Add the HBS/DNA solution dropwise into the Phoenix E culture dish and then rock the plate gently a few times to distribute DNA/CaPO4 particles evenly. Transfer the plate to a 37°C incubator.
- Twenty-four hours posttransfection, change the culture medium. Gently add 6 ml of fresh medium per 10 cm plate. Leave cells overnight in a 32°C incubator; retroviruses are more stable at 32°C than at 37°C. In addition, seed 2 × 105 AM12 cells per 10cm plate in 10ml medium and incubate overnight at 37°C.
- The next day (i.e., 48 h posttransfection) harvest virus by collecting the medium from Phoenix E cultures. Gently add fresh medium to cells and harvest virus again 8h later. Add fresh medium for a third time, incubate the cells overnight at 32°C and collect the medium. All three supernatants contain active virus. Each viral supernatant should be filtered using a 0.45-µm filter on collection and either used immediately (i.e., added to AM12 cells) or stored at-80°C (titre will halve on each freeze/thaw cycle).
- Virus harvested from Phoenix E cells is used to infect AM12 cells. Remove 8ml medium from the AM12 culture. Add 5 µl polybrene (5mg/ml) and 3ml viral supernatant to each plate (5ml total volume), shake gently, and place in a 32°C incubator for 24 h.
- Twenty-four to 48 h postinfection the AM12 cells are 60-80% confluent and are ready to be selected by adding 2µg/ml puromycin to the medium. Change medium every 2-3 days. Once the cells are confluent, passage them, prepare frozen cell stocks, and use the rest to infect keratinocytes.
- For some applications, AM12 are subjected to further selection to achieve maximal viral titres. This can be achieved by either cloning at limiting dilution or, if there is a suitable surface marker, FACS sorting of bulk populations. Both methods have been described previously (Levy et al., 1998).
H. Retroviral Infection of Cultured Human Keratinocytes Using Viral Supernatant
J2 puro are 3T3 J2 cells that have been stably transfected with pBabe puro to render them resistant to puromycin (Levy et al., 1998). They are handled in the same way as 3T3 J2 cells, except that the medium is supplemented with 2 µg/ml puromycin.
Steps
- Grow infected AM12 packaging cells to 95% confluence and transfer to keratinocyte culture medium. Collect virus over a 24- to 48-h period at 32°C, harvesting virus every 8-12 h and replacing with fresh medium. The virus should be filtered through a 0.45-gm filter on collection and either used immediately or stored at -80°C (titre will halve on each freeze/thaw cycle).
- Keratinocytes with the greatest proliferative potential (putative stem cells) can be enriched by rapid adhesion to collagen (Jones and Watt, 1993). Selection of keratinocytes on collagen prior to infection increases the retroviral infection efficiency to 60-80%. Harvest subconfluent keratinocytes, first removing the 3T3 J2 feeders. Plate 2 × 106 keratinocytes per 10-cm collagencoated plate (or equivalent plating density if smaller dishes are used). Allow cells to adhere for 15-20min. Wash gently with PBS and add fresh keratinocyte medium and mitomycin C-treated J2 puro cells. The final volume of medium should be 10 ml per 10-cm dish. Incubate cells overnight at 37°C.
- The next day, remove 8 ml of medium and add 3ml of retrovirus supernatant and 5gl polybrene (5 mg/ml).
- Incubate cells overnight at 32°C. The next day remove virus containing medium and add fresh keratinocyte medium.
- Infected keratinocytes are ready to use or can be transferred to puromycin containing medium and passaged 24h later.
Retroviral Infection of Cultured Human Keratinocytes by Coculture with Packaging Line
Steps
- Prepare AM12 by treatment with mitomycin C in the same way as for 3T3 J2 cells (Section III,A).
- Plate keratinocytes in the same way as when plating on 3T3 J2 cells (Section III,E). Use complete keratinocyte medium.
- After 2-3 days remove AM12, with EDTA treament, as for 3T3 J2 cells (Section III,E). Add mitomycin C-treated J2 puro and supplement the culture medium with puromycin.
IV. COMMENTS
It is possible to obtain up to 100 population doublings of neonatal foreskin keratinocytes prior to senescence (Rheinwald, 1989). The number of passages prior to senescence varies between cell strains: 5 is the minimum, 10 is the average, and greater than 20 is the maximum.
The same basic culture procedure can be used to grow keratinocytes from adult epidermis (although the number of population doublings obtained will be somewhat reduced) and from other stratified squamous epithelia, such as the lining of the mouth and the exocervix (Rheinwald, 1989).
Methods for transfecting Phoenix cells and retrovirally infecting AM12 cells are based on those of Swift et al. (1999; http://www.stanford.edu/group/nolan/ index.html). We routinely use the pBabe puro retroviral vector (Morgenstern and Land, 1990). Puromycin is the optimal drug for selection, as it is not toxic to transduced keratinocytes and kills nontransduced cells within 2-4 days (Levy et al., 1998). Following culture and selection, the proportion of stably transduced cells obtained with the supernatant and coculture methods are equal (>90%). The advantage of the supernatant method is that large numbers of cells are infected simultaneously; however, with the coculture method, fewer keratinocytes are required initially.
V. PITFALLS
- Under the conditions described, fibroblast contamination of keratinocytes is very rarely a problem because the feeder layer and culture medium suppress the growth of any human fibroblasts isolated from the skin at the same time as the keratinocytes (Rheinwald, 1975). We have noted that some strains of keratinocytes contain ndk-like (for nondifferentiating keratinocyte; see Adams and Watt, 1988) epithelial cells, but these cells are not abundant and can usually be removed with EDTA.
- When keratinocytes are plated in culture there is selective attachment of the basal cells, but within 1 day the cultures consist of a mixture of basal and terminally differentiating keratinocytes (e.g., Jones and Watt, 1993). Seeding keratinocytes at high density appears, in our experience, to promote terminal differentiation and is not, therefore, the answer if you are in a hurry to obtain more cells.
- Because keratinocytes are maintained in culture for long periods it is essential to be scrupulous in sterile technique and laboratory cleanliness to avoid fungal or bacterial contamination. We spray our incubators three times a week with Mikrozid.
- It is essential to be well organised and to plan your experiments in advance. It takes about 10 days from plating for keratinocytes to be ready for use and sufficient feeders must be available on the days when keratinocytes are ready for passaging.
- Use of an amphotropic retrovirus is potentially hazardous because the virus can infect human cells. Special care should therefore be taken and appropriate local safety guidelines followed.
References
Adams, J. C., and Watt, E M. (1988). An unusual strain of human keratinocytes which do not stratify or undergo terminal differentiation in culture. J. Cell Biol. 107, 1927-1938.
Compton, C. C., Gill, J. M., Bradford, D. A., Regauer, S., Gallico, G. G., and O'Connor, N. E. (1989). Skin regenerated from cultured epithelial autografts on full-thickness burn wounds from 6 days to 5 years after grafting: A light, electron microscopic and immunohistochemical study. Lab. Invest. 60, 600-612.
Dajee, M., Lazarov, M., Zhang, J. Y., Cai, T., Green, C. L., Russell, A. J., Marinkovich, M. P., Tao, S., Lin, Q., Kubo, Y., and Khavari, P. A. (2003). NF-KB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 421, 639-643.
Dellambra E., Prislei, S., Salvati, A. L., Madeddu, M. L., Golisano, O., Siviero, E., Bondanza, S., Cicuzza, S., Orecchia, A., Giancotti, E G., Zambruno, G., and De Luca, M. (2001). Gene correction of integrin β4-dependent pyloric atresia-junctional epidermolysis bullosa keratinocytes establishes a role for β4 tyrosines 1422 and 1440 in hemidesmosome assembly. J Biol Chem. 276, 41336-41342.
Gerrard, A. J., Hudson, D. L., Brownlee, G. G., and Watt, E M. (1993). Towards gene therapy for haemophilia B using primary human keratinocytes. Nature Genet. 3, 180-183.
Jones, P. H., and Watt, E M. (1993). Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell 73, 713-724.
Levy, L., Broad, S., Diekmann, D., Evans, R. D., and Watt, E M. (2000). β1 integrins regulate keratinocyte adhesion and differentiation by distinct mechanisms. Mol. Biol. Cell. 11, 453-466.
Levy, L., Broad, S., Zhu, A. J., Carroll, J. M., Khazaal, I., P6ault, B, and Watt, E M. (1998). Optimised retroviral infection of human epidermal keratinocytes: Long-term expression of transduced integrin gene following grafting on to SCID mice. Gene Ther. 5, 913-922.
Markowitz, D., Goff, S., and Bank, A. (1988). Construction and use of a safe and efficient amphotropic packaging cell line. Virology. 167, 400-406.
Morgenstern, J. P., and Land, H. (1990). Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587-3596.
Ortiz-Urda, S., Thyagarajan, B., Keene, D. R., Lin, Q., Fang, M., Calos, M. P., and Khavari, P. A. (2002). Stable nonviral genetic correction of inherited human skin disease. Nature Med. 8, 1166-1170.
Rheinwald, J. G. (1989). Methods for clonal growth and serial cultivation of normal human epidermal keratinocytes and mesothelial cells. In "Cell Growth and Division: A Practical Approach" (R. Baserga, ed.) pp. 81-94. IRL Press, Oxford.
Rheinwald, J. G., and Green, H. (1975). Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinizing colonies from single cells. Cell 6, 331-343.
Ronfard, V., Rives, J. M., Neveux, Y., Carsin, H., and Barrandon, Y. (2000). Long-term regeneration of human epidermis on third degree burns transplanted with autologous cultured epithelium grown on a fibrin matrix. Transplantation 15, 1588-1598.
Swift, S. E., Lorens, J. B., Achacoso, P., and Nolan, G. P. (1999). Rapid production of retrovirus for efficient gene delivery to mammalian cells using 293T cell-based systems. In "Current Protocols in Immunology" (J. E. Coligan, A. M. Kruisbeek, D. H. Margulies, E. M. Shevach, and W. Strober, eds.), Vol 1.17C, pp: 1-17. Wiley, New York.
Zhu, A. J., and Watt, E M. (1996). Expression of a dominant negative cadherin mutant inhibits proliferation and stimulates terminal differentiation of human epidermal keratinocytes. J. Cell Sci. 109, 3013-3023.
Zhu, A. J., and Watt, E M. (1999). β-Catenin signalling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development 126, 2285- 2298
Support our developers




