Detection of Cell Cycle Stages in situ in Growing Cell Populations
I. INTRODUCTIONMicroscopic in situ detection of the cell cycle stages is based on immunocytochemical techniques and allows one to determine the stage of the cell cycle of individual cells. Of numerous antigenes specific for a certain cell cycle stage, the most often used one is nuclear protein Ki-67 (pKi-67). This protein is expressed only in cycling cells and is, therefore, used routinely in histopathological analyses to assess proliferating activity in suspected neoplastic tissues. Despite many investigations, its function remains not wholly clear (Endl and Gerdes, 2000). Importantly, pKi-67 changes its intranuclear distribution during progression of the cell cycle (Bridger et al., 1998; Kill, 1996; Starborg et al., 1996). Figure 1 shows the distribution of pKi-67 at the main cell cycle stages of human primary fibroblasts. In early G1, pKi-67 accumulates in the assembling nucleoli and in sites enriched in heterochromatin; it forms numerous granules scattered over the nucleoplasm ("jaguar" pattern). Thenmin late G1, during the entire S phase, and in G2mpKi-67 is localized predominantly in the nucleoli, mostly at their periphery. During late G2 and early prophase, when the nucleoli disassemble, the protein is diffusely distributed in the nucleoplasm. In later prophase, pKi-67 coats the surface of condensing chromosomes (forming "perichromosomal layer") and persists on the surface of mitotic chromosomes until late telophase. The protein especially strongly decorates heterochromatin regions of mitotic chromosomes (with exception to the centromeric heterochromatin), while NORs are free from pKi-67 (Traut et al., 2002). Biochemical studies indicate that pKi-67 resides in the regions of the nucleus containing densely packed chromatin, most probably, heterochromatin (Kreitz et al., 2000), and presumably, is involved in chromatin compaction. Indeed, the C-terminal domain of pKi-67 binds mammalian heterochromatin protein 1 (HP1) (Scholzen et al., 2002) and sites where pKi-67 accumulates in early G1 become later HPl-binding foci (Kametaka et al., 2002).
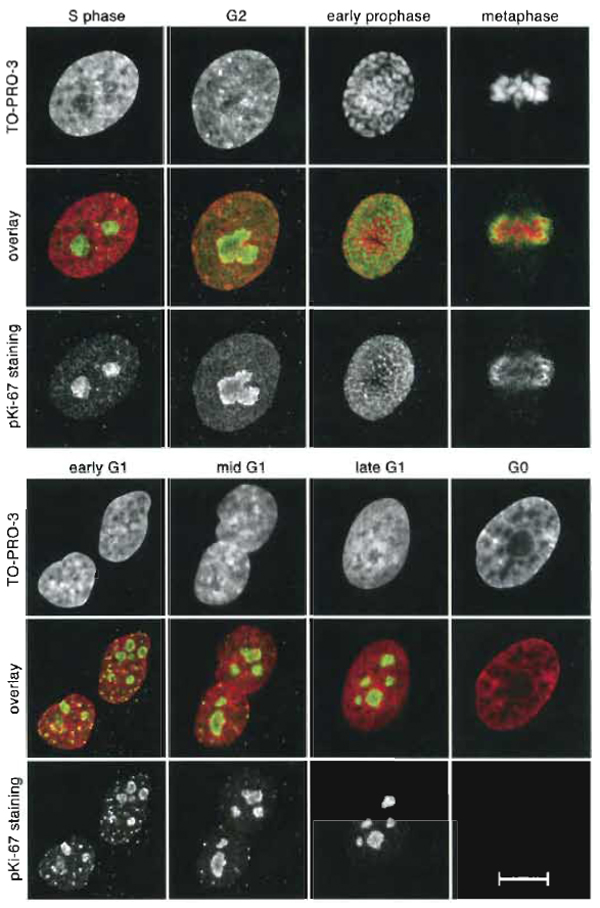 |
| FIGURE 1 Immunostaining of pKi-67 (protocol A). The cells stained are human primary fibroblasts. For each stage of the cell cycle, three images represent DNA staining with TO-PRO-3 (top), pKi-67 immunostaining (bottom), and an overlay of the two with pseudocolors (mid): red for TO-PRO-3 and green for pKi-67. Each image is a maximum intensity projection of four midconfocal sections (1 µm) through a nucleus. S phase (S-phase cells were identified by BrdU labeling, data not shown): only nucleoli are stained; a weak diffuse staining is observed in the nucleoplasm. G2: nucleoli often fuse, forming one huge nucleolus of a lobulated shape with an irregular edge; staining of the nucleoplasm is more intense than in S phase. Early prophase: pKi-67 is distributed over the entire nucleus and occupies all the space between condensing chromosomes. Metaphase: protein covers condensed chromosomes. The same distribution is observed in later prophase and in anaphase (not shown). Early GI: characteristic "jaguar" staining pattern; pKi-67 is located in reassembling nucleoli and in small multiple foci in the nucleoplasm. Mid GI: the number of foci in the nucleoplasm decreases. Late GI: staining is limited to the nucleoli. GO: in contrast to cycling cells, quiescent cells do not express pKi-67. Note that in cycling cells (but not in GO cells), small foci of pKi-67 are also present in the cytoplasm; apparently, they are sites of pKi-67 synthesis. Scale bar: 10µm. |
BrdU labeling also allows one to differentiate among early, mid, and late S-phase stages (Fig. 2). Pulse labeling with two halogenated nucleotides during two different time points results in differential staining of chromatin replicating at different periods of S phase in the same nuclei (Aten et al., 1992, 1993, 1994) (protocol D). Double replication labeling is an indispensable method used to establish the time sequence of replication events and is widely used in the studies of DNA replication in different cells and organisms (e.g., Ma et al., 1998; Visser et al., 1998; Zink et al., 1999; Habermann et al., 2001; Alexandrova et al., 2004). To exemplify the application of this method, Fig. 3 shows different nuclear location of early (green) and mid (red) replicating chromatin in human fibroblast nucleus visualized by double replication labeling.
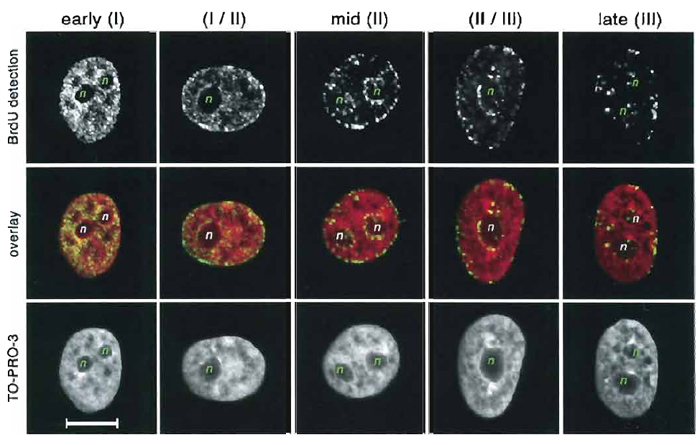 |
| FIGURE 2 Replication labeling with BrdU (protocol C). Neuroblastoma SH-EP N14 cells were pulse
labeled with BrdU for 40min; BrdU immunodetection was performed using DNase digestion, counterstaining
with TO-PRO-3. For each stage of the S phase, three images represent BrdU labeling (top), DNA staining
with TO-PRO-3 (bottom), and overlay of the two with pseudocolors (mid): red for TO-PRO-3 and green for
BrdU. Each image is a maximum intensity projection of four midconfocal sections (1 µm) through a nucleus.
Three characteristic patterns change one another as the S phase proceeds. Early S phase (pattern I): all nucleoplasm
is labeled with exception to the nucleoli (n) and speckles (dark areas in counterstaining). Mid S phase
(pattern II): characteristic labeling is seen along the nuclear periphery and the border of the nucleolus (n);
between these sites, signals are rare or lacking. Late S phase (pattern III): replication foci are significantly
larger than in early and mid S phase; signals are observed at the periphery of the nucleus, at the border of
nucleoli (n), and in nucleoplasm between them. Two transient patterns (I/II and II/III) are sometimes considered as separate S-phase stages (O'Keefe et al., 1992). Scale bar: 10µm. |
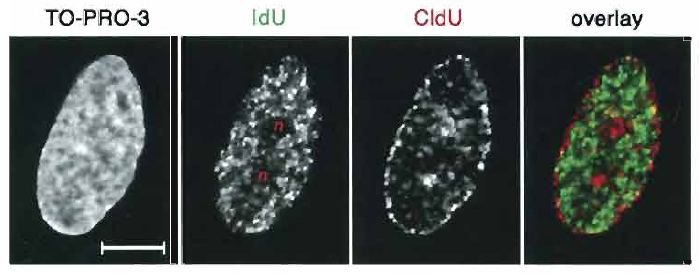 |
| FIGURE 3 Double replication labeling with IdU and CIdU (protocol D). Primary human fibroblasts were pulse labeled with IdU (green) in early S phase and with CldU (red) in mid S phase after a 4-h chase. Counterstaining with TO-PRO-3. Images represent the same midoptical section through the nucleus. Note that the midreplicating chromatin is mainly located at the very nuclear periphery and around nucleoli (n), whereas early replicating chromatin is distributed throughout the nuclear interior with exception of the nucleoli. Scale bar: 10 µm. |
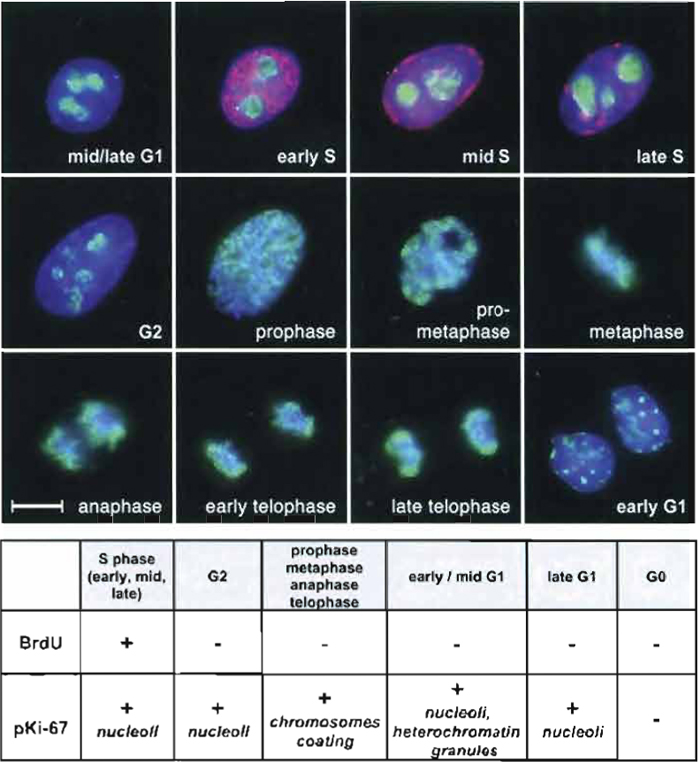 |
| FIGURE 4 Combined immunodetection of pKi-67 and BrdU (protocol E). Neuroblastoma cells SH-EP
N14 were pulse labeled with BrdU for 30 min. After simultaneous detection of pKi-67 (green) and BrdU (red),
nuclei were counterstained with DAPI (blue). Images are overlays of three gray scale pictures (one for each
fluorochrome) collected using an epifluorescence microscope. Differences among stages of the cell cycle are
summarized in Table I. Scale bar: 10µm. |
A combination of pKi-67 immunostaining and BrdU immunodetection (protocol E; Fig. 4) allows one to differentiate between all cell cycle stages with exception to late G1 and G2. It may be noted though that G2 nuclei have a more diffuse distribution of pKi-67 and fusing nucleoli; they are also noticeably larger than G1 nuclei. Though these characters are not clearly manifested in all cell types, with some experience they often help distinguish between G1 and G2 cells.
It is noteworthy, for a growing cell population with a known doubling time, that combined pKi-67 staining and BrdU labeling are sufficient to estimate duration of the main cell cycle stages. Duration of the cell cycle (or doubling time), Td, is calculated from the formula:
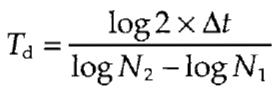 |
where N1 and N2 are the number of counted cells per given area of a coverslip at time points 1 and 2, correspondingly, and Δt is the duration between two observations. The doubling time is equivalent to the cell cycle length when 100% of the cells proliferate, as can be estimated by immunodetection of pKi-67.
The proportion of quiescent (G0) cells, pG0, is simply the proportion of pKi-67 negative cells. The length of S phase (Ts) is proportional to the proportion of Sphase (ps, BrdU positive) cells:
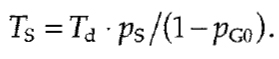 |
Similarly, the lengths of each substage of S phase can be estimated from proportions of cells showing early, mid, and late replication patterns, correspondingly The proportion of early G1 cells, PG1e, can be estimated by counting nuclei with specific "jaguar" pattern and duration of early G1:
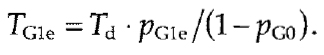 |
The length of mitosis and G2 do not vary significantly in mammalian cells and it is therefore assumed that they take about 1 and 2-3 h, respectively. On this basis, one can also assess the total duration of G1, TG1, as difference between doubling time and duration of other cell cycle stages:
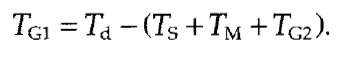 |
These calculations are simplified for immortalized cell lines in which there are no quiescent cells, i.e., essentially all cells are cycling, as, e.g., HeLa cells. Such populations indeed show a nearly 100% positive staining with pKi-67. In such cases,
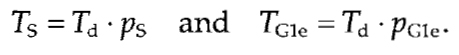 |
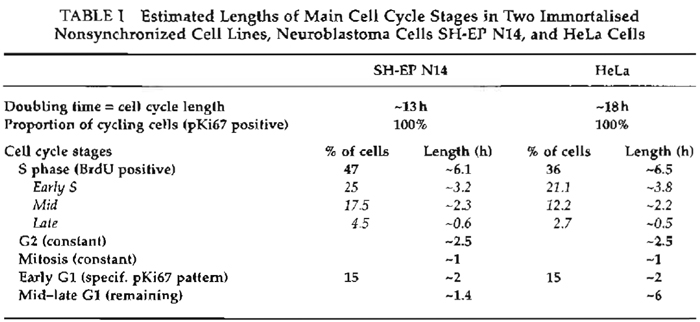
Table I exemplifies estimation of cell cycle stages lengths for two immortalized cell lines: neuroblastoma cells SH-EP N14 and HeLa.
II. MATERIAL AND INSTRUMENTATION
- Grow cell on coverslips of desirable size and thickness of 0.17 ± 0.01 mm. In case of cells growing in suspension, we recommend attaching living cells to coverslips coated with polylysine as described in detail elsewhere (Solovei et al., 2002b).
- We recommend performing fixation and washings in 6-well plates; each well accommodates one coverslip of size ranging from 15 × 15 to 22 × 22.
- To handle coverslips, use fine forceps (e.g., Dumont No. 4 or 5).
- Special care must be taken not to dry cells at any steps of immunostaining. Make sure that when solutions are changed, coverslips always remain covered with liquid.
- For incubation with antibody, use humid dark chambers with Parafilm on the bottom. Place drops of antibody solutions on Parafilm, and lay coverslips with cells on these drops with the cell side down. Carry out incubations at room temperature.
- We recommend using Vectashield (Vector Laboratories; Cat. No. H-1000) as the antifade mounting medium. To mount cells, place a drop of Vectashield on a clean microscopic slide and quickly put coverslip with cells upside down on the antifade. Gently remove excess antifade medium with soft paper and seal with colourless nail polish (preferably, so called, base coat).
- Mounted and sealed preparations can be stored in the dark at 4°C for months.
III. PROCEDURES
A. Immunostaining with Anti-Ki67 Antibodies
Solutions
- Cell culture mediums used for respective cells
- Phosphate-buffered saline (l× PBS): 140mM NaCl, 2.7mM KCl, 6.5 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.2
- Fixative: 3.7% formaldehyde made before use from 37% stock solution (Sigma, Cat. No. F-1268) by dilution with 1× PBS
- PBST: PBS with 0.05% Tween 20 (Calbiochem, Cat. No. 655204)
- 1× PBS with 0.04% sodium azide (Merck, Cat. No. 106688)
- 0.5% Triton X-100 (Sigma, Cat. No. T-8787) in 1× PBS
- Blocking solution: 4% bovine serum albumin (BSA) in PBST. Store the stock solution of 12% BSA in PBS with 0.04% sodium azide at 4°C
- Mouse anti-pKi-67 (DakoCytomation, Cat. No. M7240); working dilution- 1" 100 in blocking solution
- Any antimouse antibodies conjugated to a desirable fluorochrome. As two examples, we suggest goat antimouse-Cy3 (orange-red fluorescence, Jackson ImmunoResearch Laboratories, Cat. No. 115-165-072; working dilution- 1 "200) or goat antimouse-Alexa 488 (green fluorescence; Molecular Probes, Cat. No. A- 11017; working dilution- 1"500); antibodies are diluted in blocking solution
- DNA-specific dye. The choice depends on the available microscopic filters or confocal laser lines and on fluorochrome chosen for secondary antibodies. The most popular counterstaining dyes are 4',6-diamidino- 2-phenylindole (DAPI, Sigma, Cat. No. D-9564) with blue fluorescence; propidium iodide (PI, Sigma, Cat. No. P-4170) with red fluoroscence; and TO-PRO-3 (Molecular Probes, Cat. No. T-3605) with emission in the far-red part of the spectrum. DAPI stock = 5 µg/ml in H2O, working solution- 0.05µg/ml. PI stock- 500µg/ml in H2O, working solution- 25µg/ml. TOPRO- 3 stock- 1 mM in dimethyl sulfoxide (DMSO); working solution - 1 µM. All stock solutions are kept frozen; working dilutions in PBST are done before use
- RNase (Roche, Cat. No. 109169); stock solution = 20mg/ml in 10mM Tris-HCl + 15mM NaCl; working solution- 0.2mg/ml in PBS
Steps
- Fix cells in 3.7% formaldehyde/PBS at room temperature for 10 min; wash in PBST, 3 × 5 min. If needed, fixed cells can be kept in PBS with 0.04% sodium azide at 4°C overnight or longer.
- Permeabilize with 0.5% Triton X-100 in PBS for 5 min.
- Incubate in blocking solution for 10min.
- Incubate with primary antibodies, mouse antipKi- 67 for 30min and wash with PBST 3 × 5 min.
- Incubate with secondary antibodies, e.g., goat antimouse Alexa488 or goat antimouse-Cy3, for 30min and wash with PBST 3 × 5 min.
- Counterstain nuclei with DNA-specific dye (DAPI, PI, or TO-PRO-3) for 5min and then rinse briefly in PBST. Optional: Because PI and TO-PRO-3 also stain RNA, slides can be preincubated in RNase working solution at 37°C for 30min.
- Mount cells in antifade medium and seal with nail polish.
B. Replication Labeling with BrdU and Detection with Heat Denaturation
Solutions
- 5-bromo-2'-deoxyuridine (Sigma, Cat. No. B-9285). Make a 1 mM stock solution in H2O, store aliquots at -20°C
- 2 × SSC, pH 7.0; 1 × SSC is 0.15 M NaCl and 0.015 M sodium Na-citrate
- 0.1N HCl
- 50% formamide (Merck, Cat. No. 1.09684) in 2× SSC. Formamide is toxic, so all steps that involve the use of this reagent should be performed in a hood and gloves should be worn.
- 70% formamide in 2 × SSC.
- Mouse anti-BrdU (Roche, Cat. No. 7580), working dilution = 1:200 in blocking solution.
- Any antimouse antibodies coupled to desirable fluorochrome. As two examples, we suggest the same secondary antibodies as for detection in protocol A.
The rest of the solutions are the same as for protocol A.
Steps
- Grow cells to ≥50% confluency.
- Add BrdU to the cell culture medium to a final concentration of 10-20µM and incubate for 15-60 min at 37°C in a CO2 incubator.
- Fix and permeabilize cells as described in protocol A.
- Incubate in 0.1N HCl for 10min.
- Wash with 2 × SSC for 5 min and equilibrate in 50% formamide/2 × SSC at room temperature for a minimum of 15min. At this step, slides may be kept in 50% formamide at 4°C overnight or longer.
- For cell DNA denaturation, warm up 70% formamide/ 2 × SSC to 70°C immerse coverslips with cells for 2min, and then immediately move them to ice-cold PBST and wash with PBST 3 × 5 min.
- Incubate in blocking solution for 10min.
- Incubate with primary antibodies, mouse anti- BrdU for 30min and wash with PBST 3 × 5 min.
- Incubate with secondary antibodies, e.g., goat antimouse Alexa488 or sheep antimouse-Cy3, for 30 min and wash with PBST 3 × 5 min.
- Mount cells in antifade medium and seal with nail polish.
Comments
BrdU pulse length can vary: only 5rain may be sufficient to detect the typical replication pattern. However, extended incubation time results in brighter signals, as more BrdU per replication focus is incorporated. At the same time, the replication pattern gradually becomes less distinct as pulse length exceeds 1 h.
C. Replication Labeling with BrdU and Detection with DNase Digestion
Roche Applied Science sells a kit for BrdU detection for immunofluorescence microscopy (Cat. No. 1296736), which is based on enzymatic digestion of DNA. The antibody to BrdU supplied with the kit contains nucleases generating single-stranded DNA fragments in the nucleus. As an alternative to the kit, this section describes a simple and inexpensive method that allows incorporated molecules of BrdU to be exposed to antibodies after DNase digestion.
Solutions
- DNase I (Roche, Cat. No. 104 159); to make a stock solution, dilute to 1 mg/ml in 0.15M NaCl in 50% glycerol, make aliquots, and store them at-20°C.
- DNase incubation buffer: 0.5 × PBS, 30mM Tris, 0.3mM MgCl2, 0.5mM 2-mercaptoethanol, 0.5% bovine serum albumin, and 10µg/ml DNase I.
The rest of the solutions are the same as for protocol A.
Steps
- Fix cells in 3.7% formaldehyde/PBS at room temperature for 10 min; wash in PBST, 3 × 5 min. If needed, fixed cells can be kept in PBS with 0.04% sodium azide at 4°C overnight or longer.
- Permeabilize with 0.5% Triton X-100 in PBS for 5 min.
- Incubate in blocking solution for 10min.
- Incubate in blocking solution for 10min.
- Dissolve primary antibodies against BrdU in the DNase incubation buffer and incubate as specified in protocol B.
The rest of the steps are the same as steps 9 and 10 in protocol B.
Pitfalls
The most probable cause for weak staining is bad DNase. In addition, an extra step with incubation in 0.1N HCl for 5-10min (see step 4 in protocol B) is recommended.
D. Double Labeling with IdU and CldU
Solutions
- 5-Iodo-2'-deoxyuridine (IdU) (Sigma, Cat. No. 1- 7125). Make a 1 mM stock solution in 40% DMSO, store aliquots at-20°C
- 5-Chloro-2'-deoxyuridine (CldU) (Sigma, Cat. No. C-6891). Make a 1 mM stock solution in H2O, store aliquots at-20°C
- Mouse anti-IdU and -BrdU [Becton Dickinson; Cat. No. 347580 (7580), clone B44], working dilution- 1 :50
- Goat antimouse-Alexa488 highly cross-adsorbed (Molecular Probes; Cat. No. A-11029), working dilution = 1 "500
- Rat anti-CldU and -BrdU [Harlan Sera-Labs, Cat. No. MAS 250, clone BU1/75 (ICR1)], working dilution = 1:200
- Donkey antirat-Cy3 (Jackson ImmunoResearch, Cat. No. 712-165-153), working dilution - 1:200. All antibodies are diluted in blocking solution. Of course one can use a different pair of fluorochromes conjugated to secondary antibodies than Alexa488 and Cy3. Secondary antibodies themselves can also be changed without serious risk to spoil staining, whereas the choice of primary antibodies is crucial for successful staining.
- High stringency buffer: 0.5M NaCl, 36 mM Tris, 0.5% Tween 20, pH 8.0
The rest of the solutions are the same as for protocol A.
Steps
- Grow cells to ≥50% confluency.
- . Add the required amount of IdU to the cell culture medium at a final concentration of 1-10µM; incubate for 30-45 min.
- Remove incubation medium, rinse cells twice in PBS, and replace with fresh medium. Chase: incubate cells in fresh medium for the period equal to one-half to three-fourth of the S-phase duration. A chase time of 4-6 h is recommended for, e.g., human and mouse fibroblasts, HeLa cells, stimulated human lymphocytes, and lymphoblastoid cells.
- Add the required amount of CldU to the cell culture at a final concentration of 1-10 µM; incubate for 15 to 45 min.
- Fix cells in 3.7% formaldehyde/PBS at room temperature for 10min; wash in PBST, 3 × 5min. If necessary, fixed cells could be kept in PBS with 0.04% sodium azide at 4°C overnight or longer.
- Permeabilize cells with 0.5% Triton X-100 in PBS for 5 min.
- The denaturation of DNA can be done either by heating in formamide (steps 5-7 in protocol B) or by DNase digestion (as described in protocol C). In the latter case, antibodies diluted in DNase incubation buffer should be rat anti-CldU/-BrdU (see later).
- The recommended rat anti-BrdU antibodies detect both BrdU and CldU, but not IdU, and should be applied first. The recommended mouse anti-BrdU antibodies (Becton Dickinson), which are used for the detection of IdU, also have some affinity to CldU and must therefore be used after the detection of CldU. Apply antibodies against CldU (rat anti-CldU and -BrdU), incubate for 30min, and wash in PBST 3 × 5 min.
- Apply secondary antibodies for CldU detection, donkey antirat-Cy3; incubate for 30min and wash in PBST 3 × 5 min.
- Apply antibodies against IdU (mouse anti- IdU/-BrdU), incubate for 30 min, and wash in PBT 3 × 5 min.
- Wash in the stringency buffer for 10min to remove nonspecifically bound antibodies. This step is important, as some binding of mouse anti-IdU antibodies to CldU epitops still takes place, even after primary rat anti-CldU and secondary goat antirat antibodies were applied.
- Apply secondary antibodies for IdU detection, goat antimouse-Alexa488, incubate in a dark humid chamber for 30min, and wash in PBST 3 × 5 min.
- Counterstain nuclei (with DAPI and/or TOPRO- 3), mount cells in antifade medium, and seal with nail polish.
E. Combination of Both Methods with Anti-Ki-67 Staining
Epitops of Ki-67 protein are stable and the protein can be detected after 0.1N HCl and denaturation steps (with heat denaturation or DNase). Therefore, pKi-67 immunostaining can be performed either simultaneously or after detection of incorporated halogenated thymidine analogues. For simultaneous detection, primary antibodies against pKi-67 and BrdU are mixed; both secondary antibodies may also be applied as one solution.
Solutions
- Mouse anti-BrdU (Roche, Cat. No. 7580); working dilution = 1:200
- Rabbit anti-Ki-67 (DakoCytomation, Cat. No. A0047); working dilution = 1:50
- Goat antimouse-Cy3 (Jackson ImmunoResearch Laboratories, Cat. No. 115-165-072); working dilution = 1:200
- Goat antirabbit-FITC (BioSource, Cat. No. ALI 4408); working dilution = 1:200
All antibody dilutions are done in blocking solution.
Steps
- Grow cells to ≥50% confluency.
- Add BrdU to the cell culture medium to a final concentration of 10-20µM and incubate for 15-60 min at 37°C in a CO2 incubator.
- Fix and permeabilize cells as described in protocol A.
- Incubate in 0.1N HCl for 10min.
- Wash with 2 × SSC for 5 min and equilibrate in 50% formamide/2 × SSC at room temperature for a minimum of 15min. At this step, slides may be kept in 50% formamide at 4°C overnight or longer.
- For cell DNA denaturation, warm up 70% formamide/ 2 × SSC to 70°C immerse coverslips with cells for 2min, and then immediately move them to ice-cold PBST and wash with PBST 3 × 5 min.
- Incubate in blocking solution for 10min.
- Incubate in mixture of mouse anti-BrdU and rabbit anti-Ki-67 antibodies at room temperature and wash with PBST 3 × 5 min.
- Incubate in a mixture of goat antimouse-Cy3 and goat antirabbit-FITC and wash with PBST 3 × 5 min.
- Counterstain nuclei, mount cells in antifade medium, and seal with nail polish.
Pitfalls
Special care should be taken to prevent crossreaction of antibodies: use preadsorbed secondary antibodies and avoid using in the same experiment primary antibodies raised in closely related animals, e.g., mouse and rat or sheep and goat.
References
Alexandrova, O., Solovei, I., Cremer, T., and David, C. (2003). Replication labeling patterns and chromosome territories typical of mammalian nuclei are conserved in the early metazoan Hydra. Chromosoma 112, 190-200.
Aten, J. A., Bakker, R J., Stap, J., Boschman, G. A., and Veenhof, C. H. (1992). DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem. 1. 24, 251-259.
Aten, J. A., Stap, J., Hoebe, R., and Bakker, P. J. (1994). Application and detection of IdUrd and CldUrd as two independent cellcycle markers. Methods Cell Biol. 41, 317-326.
Aten, J. A., Stap, J., Manders, E. M., and Bakker, P. J. (1993). Progression of DNA replication in cell nuclei and changes in cell proliferation investigated by DNA double-labelling with IdUrd and CldUrd. Eur. J. Histochem. 37(Suppl. 4), 65-71.
Bridger, J. M., Kill, I. R., and Lichter, P. (1998). Association of pKi-67 with satellite DNA of the human genome in early G1 cells. Chromosome Res. 6, 13-24.
Endl, E., and Gerdes, J. (2000). The Ki-67 protein: Fascinating forms and an unknown function. Exp. Cell Res. 257, 231-237.
Gratzner, H. G. (1982). Monoclonal antibody to 5-bromo- and 5- iododeoxyuridine: A new reagent for detection of DNA replication. Science 218, 474-475.
Habermann, E, Cremer, M., Walter, J., Hase, J., Bauer, K., Wienberg, J., Cremer, C., Cremer, T., and Solovei, I. (2001). Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res. 9, 569-584.
Kametaka, A., Takagi, M., Hayakawa, T., Haraguchi, T., Hiraoka, Y., and Yoneda, Y. (2002). Interaction of the chromatin compactioninducing domain (LR domain) of Ki-67 antigen with HP1 proteins. Genes Cells 7, 1231-1242.
Kill, I. R. (1996). Localisation of the Ki-67 antigen within the nucleolus: Evidence for a fibrillarin-deficient region of the dense fibrillar component. J. Cell Sci. 109, 1253-1263.
Kreitz, S., Fackelmayer, E O., Gerdes, J., and Knippers, R. (2000). The proliferation-specific human Ki-67 protein is a constituent of compact chromatin. Exp. Cell Res. 261, 284-292.
Ma, H., Samarabandu, J., Devdhar, R. S., Acharya, R., Cheng, P. C., Meng, C., and Berezney, R. (1998). Spatial and temporal dynamics of DNA replication sites in mammalian cells. J. Cell Biol. 143, 1415-1425.
O'Keefe, R. T., Henderson, S. C., and Spector, D. L. (1992). Dynamic organization of DNA replication in mammalian cell nuclei: Spatially and temporally defined replication of chromosomespecific alpha-satellite DNA sequences. J Cell Biol. 116, 1095- 1110.
Schmidt, M. H., Broll R., Bruch H. P., Bogler O., and Duchrow, M. (2003). The proliferation marker pKi-67 organizes the nucleolus during the cell cycle depending on Ran and cyclin B. J. Pathol. 199, 18-27.
Schmidt, M. H., Broll, R., Bruch, H. P., and Duchrow, M. (2002). Proliferation marker pKi-67 affects the cell cycle in a self-regulated manner. J. Cell Biochem. 87, 334-341.
Scholzen, T., Endl, E., Wohlenberg, C., van der Sar, S., Cowell, I. G., Gerdes, J., and Singh, P. B. (2002). The Ki-67 protein interacts with members of the heterochromatin protein I (HP1) family: A potential role in the regulation of higher-order chromatin structure. J. Pathol. 196, 135-144.
Solovei, I., Cavallo, A., Schermelleh, L., Jaunin, E, Scasselati, C., Cmarko, D., Cremer, C., Fakan, S., and Cremer, T. (2002a). Spatial preservation of nuclear chromatin architecture during threedimensional fluorescence in situ hybridization (3D-FISH). Exp. Cell Res. 276, 10-23.
Solovei, I., Walter, J., Cremer, M., Habermann, E, Schermelleh, L., and Cremer, T. (2002b). FISH on three-dimensionally preserved nuclei. In "FISH: A Practical Approach" (B. Beatty, S. Mai, and J. Squire, eds.), pp. 119-157. Oxford Univ. Press, Oxford.
Starborg, M., Gell, K., Brundell, E., and Hoog, C. (1996). The murine Ki-67 cell proliferation antigen accumulates in the nucleolar and heterochromatic regions of interphase cells and at the periphery of the mitotic chromosomes in a process essential for cell cycle progression. J. Cell Sci. 109, 143-153.
Tashiro, S., Walter, J., Shinohara, A., Kamada, N., and Cremer, T. (2000). Rad51 accumulation at sites of DNA damage and in postreplicative chromatin. J. Cell Biol. 150, 283-291.
Traut, W., Endl, E., Garagna, S., Scholzen, T., Schwinger, E., Gerdes, J., and Winking, H. (2002). Chromatin preferences of the perichromosomal layer constituent pKi-67. Chromosome Res. 10, 685-694.
Visser, A. E., Eils, R., Jauch, A., Little, G., Bakker, P. J., Cremer, T., and Aten, J. A. (1998). Spatial distributions of early and late replicating chromatin in interphase chromosome territories. Exp. Cell Res. 243, 398-407.
Zink, D., Bornfleth, H., Visser, A., Cremer, C., and Cremer, T. (1999). Organization of early and late replicating DNA in human chromosome territories. Exp. Cell Res. 247, 176-188.
Support our developers




