Detection of Microbial and Viral Contaminants in Cell Lines
I. INTRODUCTIONThe presence of microbial contaminantsmbacteria, fungi, mycoplasma, or protozoamin cell cultures seriously compromises virtually all research or production work involving culture technology. Although many contamination events are overt and readily apparent, others are insidious and more difficult to detect. Similarly, viral infection may be obvious if cytopathogenesis is affected, but many viruses do not induce a drastic alteration in host cells and some are present in latent forms.
This article provides representative test protocols suitable for detecting most microbes and many viruses that might be expected in cell culture systems. The perspective is that of staff operating a national cell culture resource.
II. MATERIALS
The following media and reagents are from Difco: Bacto Sabouraud dextrose-broth (Cat. No: 0382-1); Bacto fluid thioglycollate medium (Cat. No. 0256-01); beef extract (Cat. No. 0131); brain-heart infusion (BHI) broth (Cat. No. 0037-016); neopeptone (Cat. No. Bl19); proteose peptone (Cat. No. 3); YM broth (Cat. No. 0711-01); nutrient broth (Cat. No. 003-01); Bacto yeast extract (Cat. No. 012701); trypsin, 1:250 Difco certified (Cat. No. 0152-15); and blood agar base (Cat. No. 0045- 01). Trypticase soy broth powder (Cat. No. 01-162), trypticase (Cat. No. Bl1770), mycoplasma broth base (Cat. No. 11458), and mycoplasma agar base (Cat. No. 11456) are from Becton-Dickinson Microbiology Systems (formerly BBL). Fresh defibrinated rabbit blood (Cat. No. 82-8614) and sheep blood are from Editek, and North American Biologicals provided Diamond's TP-S-1 broth base powder (Cat. No. 73- 9502) and Diamond's TP-S-1 vitamin solution (40X, Cat. No. 72-2315). American Hoechst is the source of bisbenzamide fluorochrome stain (Cat. No. 33217), and oil-free, dry annealed aluminum foil is from Reynolds Aluminum (Cat. No. 1235-0). Unless specified otherwise, cell culture media (various) and sera are from ATCC, Sigma, GIBCO-BRL, or Hyclone.
Template-primers poly(rA) 9 poly(dT)12-18 (Cat. No. 7878) and poly(dA)-poly(dT)12-18 (Cat. No. 7868) are from P-L Biochemicals, and [methyl-3H]thymidine triphosphate ([3H]TTP, carrier-free, specific activity 50-60Ci/mmol, 1.0mCi/ml, Cat. No. NET 221X) is from Dupont New England Nuclear. The scintillation counter fluid (Betafluor, Cat. No. LS-151) was purchased from National Diagnostics.
The following items are required in screening for c-type viruses such as HIV and HTLV:
Rneasy minikit, Qiagen (Cat. No. 74104)
SuperScript double-stranded cDNA synthesis kit, Invitrogen (Cat. No. 11917-010)
Platinum Taq DNA polymerase, Invitrogen (Cat. No. 0966034)
Sybr Green I, Molecular Probes, Inc. (Cat. No. S7563)
DNA standard and HIV and HTLV positive cell lines, ATCC (Cat. No. 53069 CRL-8993 and CRL 8543)
Standards, primers, and probes:
A pUC18 vector containing a 9.9-kb insert from HIV-standard DNA is used as standard DNA for HIV (ATCC, Manassas, VA, ATCC No. 53069). For GAPDH and HTLV, polymerase chain reaction (PCR) products containing the primer and probe sequence for both targets have been cloned into the pUC18 vector.
Primers and probes include:
| HIV | Forward primer | 5' TCCACCTATCCCAG TAGGAGAAAT 3' |
| Reverse primer | 5' GGTCCTTGTCTTAT GTCCAGAATG 3' |
|
| TaqMan probe | 5' GATTAAATAAAATAG TAAGAATGTATAGC 3' |
|
| HTLV | Forward primer | 5' CAATCACTCATACA ACCCCCAA 3' |
| Reverse primer | 5' CTGGAAAAGACAGG GTTGGG 3' |
|
| TaqMan probe | 5' TCCTCCAGGCCATG CGCAAATACTCG 3' |
In most cases, Sigma or VWR supplied general laboratory chemicals and solvents plus such items as instruments and bacteriological or cell culture glassand plasticware.
The following additional specialty items are required: Leighton tubes (Cat. No. 3393) from Costar; cellulose filters, 0.45 and 0.22µm (Cat. Nos. HATF 14250 and GSTF 14250, respectively) from Millipore; GasPak anaerobic systems (Cat. No. 60465) from Becton-Dickinson Microbiology Systems; embryonated chicken eggs from SPAFAS; egg candlers (Cat. No. C6372N-50001) from Nasco; egg drills, cutters, and moto tool (Cat. No. 9826- 00) from Cole-Parmer; stainless-steel sterilizing pans (Cat. No. 2065-5) from Orem; and adjustable microliter pipettes and Pipetman (Cat. No. P-20 D/P-200D) from Rainin. Reference microbes, cell lines, and viruses are from the American Type Culture Collection (ATCC): Pseudomonas aeruginosa (e.g., ATCC 14502), Micrococcus salivarious (e.g., ATCC 14344), Escherichia coli (e.g., ATCC 4157), Bacteroides distasonis (e.g., ATCC 8503), Penicillium notatum (e.g., ATCC 8537), Aspergillus niger (e.g., ATCC 34467), Candida albicans (e.g., ATCC 10231), influenza virus (e.g., ATCC VR-95 or VR-810), Newcastle disease virus (e.g., ATCC VR-108 or VR-109), and Rous sarcoma virus (e.g., ATCC VR-140 or VR-724). The Gen-Probe kit is available from Fisher (Cat. No. GP1591) and the mycoplasma PCR kit is from ATCC (Cat. No. 90-1001K).
III. PROCEDURES A. Bacteria and Fungi
Tests for sterility are performed routinely at ATCC on all culture media used, on cultures submitted from the community, on cultures at various stages during the accessioning process, and on all seed and distribution freezes. Pseudomonas species, micrococci, and E. coli are common bacterial isolates, whereas Penicillium, Aspergillus, and Candida species are common fungal and yeast contaminants.
1. Preparation of Media Solutions
- Sabouraud dextrose broth: Dissolve 30g dehydrated powder in 1000 ml distilled water and dispense 10-ml aliquots into each of one hundred 16 × 150-mm test tubes. Cap each tube loosely and sterilize in an autoclave for 15min at 151b pressure (121°C on slow exhaust. After removing the tubes from the autoclave, press down caps securely and store at room temperature until used. Caps of differing colors may be used to permit ready identification.
- Nutrient broth with 2% yeast extract: Dissolve 8g of nutrient broth powder plus 20g of Bacto yeast extract in 1000ml distilled water and dispense 10-ml aliquots into each of one hundred 16 × 150-mm test tubes. Cap each tube loosely, sterilize, and store as described for solution 1.
- Thioglycollate medium: Suspend 29.8 g dehydrated powder in 1000 ml distilled water in a 3-liter flask and heat to boiling to dissolve the powder completely. Dispense 10-ml aliquots of the thioglycollate medium into each of one hundred 16 × 150-mm test tubes; cap each tube loosely. Sterilize in the autoclave as described for solution 1. After removing the tubes from the autoclave, press down caps securely and store in the dark at room temperature. This medium changes color in processing. As it dissolves it turns red or gold depending on the amount of dissolved oxygen. After autoclaving it is clear and gold in color, like nutrient broth. After cooling, the top layer of medium oxidizes and the indicator in the upper portion of the tube turns pink or red. The fluid should not be used if the indicator has changed to a red color in the lower third of the tube.
- Trypticase soy broth: Suspend 30g powder in 1000ml distilled water and mix thoroughly and warm gently until solution is complete. Dispense 10-ml aliquots of die trypticase soy booth into each of one hundred 16 x 150-mm test tubes; cap each tube loosely and sterilize and store as described for solution 1.
- BHI broth: Suspend 37 g powder in 1000 ml of distilled water, dissolve completely, and dispense 10-ml aliquots of BHI into 16 × 150-mm test tubes. Cap each tube loosely. Sterilize in the autoclave for 15min at 151b pressure on slow exhaust. After autoclaving, press down caps securely, cool, and store at 4°C.
- YM broth: Dissolve 21 g powder in 1000ml distilled water and dispense 10-ml aliquots of the broth into each of one hundred 16 × 150-mm test tubes; cap each tube loosely. Sterilize and store as described for solution 1.
- Blood agar plates: Suspend 40 g blood agar base in 950ml cold distilled water and heat to boiling to dissolve the powder completely. Sterilize in the autoclave for 15min at 151b pressure on slow exhaust. When the sterile blood agar base is cooled to 50°C, add 5% (50 ml) of pretested, fresh, defibrinated rabbit blood and mix by swirling. Dispense aseptically to 9-cm plates and store at 4°C. The rabbit blood is pretested for sterility by inoculating 0.5-ml aliquots into BHI broth and YM broth and onto blood agar base plates with subsequent incubation at 25° and 37°C. Negative results in 48 to 72 h are usually sufficient to permit use of the tested fluid.
2. Examination
Steps
- Using an inverted microscope, equipped with phase-contrast optics if possible, examine cell culture vessels individually. Scrutiny should be especially vigorous in cases where large-scale production is involved.
- Check each culture first using low power. After moving the cultures to a suitable isolated area, remove aliquots of fluid from cultures that are suspect; retain these for further examination. Alternatively, autoclave and discard all such cultures.
- Prepare wet mounts using drops of the test fluids and observe under high power.
- Prepare smears, heat fix, and stain by any conventional method using filtered solutions.
- Examine under oil immersion for microbial contaminants.
- Consult Freshney (2000) for further details.
3. Inoculation and Incubation of Test Samples
Steps
- After cryopreservng stocks of cells (Hay et al., 2000), retrieve and thaw about 5% of the ampoules from liquid nitrogen or vapor storage. Pool and mix the contents of the ampoules from each cryopreserved lot using a sterile 1-ml disposable pipette. It is recommended that antibiotics not be included in media used to prepare stocks of cells for preservation. If antibiotics are used, the pooled suspension should be centrifuged at 2000g for 20min and the pellet should be resuspended in antibiotic-free medium. A series of three such washes with antibiotic-free medium prior to testing eliminates traces of antibiotics that could obscure contamination.
- From each pool, inoculate each of the following with a minimum of 0.3 ml of the test cell suspension: (a) two blood agar plates, (b) two tubes of thioglycollate broth, (c) two tubes of trypticase soy broth, (d) two tubes of BHI broth, (e) two tubes of Sabouraud broth, (f) two tubes of YM broth, and (g) two tubes of nutrient broth with 2% yeast extract.
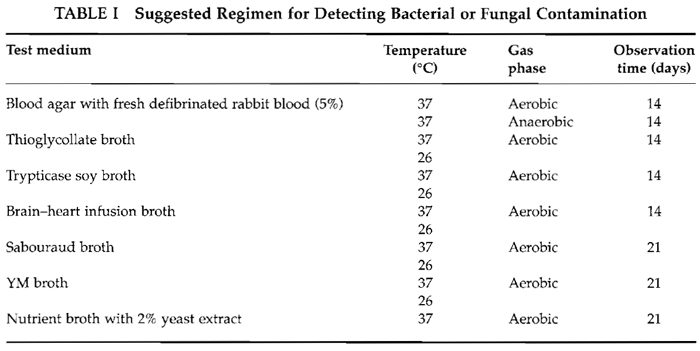
- Incubate test plates and broths as follows. (a) Blood agar plates: one at 37°C under aerobic conditions and one at 37°C anaerobically (a BBL Gaspak anaerobic system is convenient for the latter). (b) Tubes of thioglycollate broth, trypticase soy broth, BHI broth, Sabouraud broth, YM broth, and nutrient broth with yeast extract: one each at 37°C and one each at 26°C under aerobic conditions. (c) Incubate and examine periodically for 14 days the tubes of thioglycollate, trypticase soy broth, BHI broth, and blood agar plates. (d) Observe the tubes of Sabouraud broth, YM broth, and nutrient broth with yeast extract for 21 days before concluding that the test is negative. Contamination is indicated if colonies appear on solid media or if any of the liquid media become turbid.
- Repeat any components of the test series that are positive initially to confirm the presence of a contaminant.
- Autoclave and discard any contaminated cultures or ampule lots.
Comments
Of the seven media employed, trypticase soy, BHI, blood agar, and thioglycollate are suitable for detecting a wide range of bacterial contaminants. Sabouraud broth, YM broth, and nutrient broth with yeast extract will support growth of fungal contaminants. Stock media and incubation conditions used can be tested with the following ATCC control strains: P. aeruginosa, M. salivarius, E. coli, B. distasonis, P. notatum, A. niger, and C. albicans. Table I summarizes this recommended test regimen.
Pitfalls
Although this test regimen permits detection of most common bacterial and fungal organisms that grow in cell cultures, we have experiences with at least one very fastidious bacterial strain that initially escaped observation. This was present in nine different cell cultures from a single clinical laboratory in the United States submitted for testing and expansion under a government contract. The organism grew extremely slowly but could be detected after 3 weeks of incubation with cell cultures without antibiotics and with no fluid changes. Samples so developed were inoculated into sheep blood agar plates and New York City broth (ATCC medium 1685). The organism could be observed during a subsequent 6-week incubation period at 37°C
 |
The bacteriology department at ATCC determined suitable culture conditions for this microorganism and tentatively identified it as a Corynebacterium. Antibiotic sensitivity tests revealed bacteriostasis with some compounds, but no bactericidal antibiotics have yet been found.
This incident emphasizes the critical importance of diligent testing of cell cultures for contaminant microorganisms. By combining protocols such as those described here with procedures discussed later (e.g., fluorescent or nucleic acid probes for mycoplasma and viruses), one can be more certain that clean cell cultures are available for experimentation.
B. Mycoplasma
Mycoplasmal contamination of cell cultures has been established as a common occurrence that is capable of altering normal cell structure and function. Mycoplasmas have been shown to inhibit cell metabolism and growth, alter nucleic acid synthesis, affect cell antigenicity, induce chromosomal alterations, interfere with virus replication, and mimic viral actions. Basically, the growth of mycoplasma in cell cultures can be detected either by a direct microbiological agar culture procedure or by indirect procedures using staining, biochemical methods, or nucleic acid hybridization techniques (McGarrity, 1982; Hay et al., 1989, 1992, 2000).
In testing cell cultures for contamination, both direct and indirect procedures should be employed. The indirect method employed most frequently at the ATCC was originally described by Chen (1977). It requires the bisbenzamide DNA fluorochrome staining procedure plus an indicator cell. This adaptation is described here with slight modifications.
Duplicate screening techniques are generally recommended for rigorous cell line testing. Alternative methods to those included here include nucleic acid hybridization and a new technique involving the polymerase chain reaction. Details are available elsewhere (Hu and Buck, 1993; ATCC website http://www.atcc.org).
In screening cell lines for mycoplasma contamination, it is important to include positive controls in order to be assured that the test systems being used are optimal. Special precautions, however, are necessary when working with such material. The handling of mycoplasma cultures should be done at the end of a particular test and, when possible, in an isolated area using a biohazard-type hood. All equipment used in manipulations involving control cultures should be collected and sterilized immediately by autoclaving. More detailed accounts of the measures necessary to detect and prevent the spread of contamination can be found elsewhere (Uphoff and Drexler, 2001; Freshney, 2000).
1. Direct Method
The procedures used in preparing and pretesting the following culture media should be standardized, and the final pH should be adjusted to 7.2 to 7.4. Both media are prepared in quantities to be utilized within 3 to 4 weeks. The quality of the major components of the media may vary from batch to batch in the degree of toxicity and in their ability to support the growth of mycoplasma. The growth-promoting properties of each new lot of freshly prepared broth and agar media are determined by making inoculations using ATCC 23206, Acholeplasma laidlauii, and ATCC 23838, Mycoplasma arginini. In addition, the horse serum, like all sera employed for cell culture work, is screened for mycoplasmal contamination. Briefly, a 100-ml aliquot of the serum being tested is used as the serum supplement for 400ml of broth medium. The cultures are incubated aerobically at 37°C for 4 weeks and are observed for turbidity and change in pH. Subcultures to agar plates and inoculations onto Vero indicator cultures for the indirect test are performed weekly during the incubation period. In testing samples in which bacterial and/or fungal contaminations may be prevalent, penicillin and thallium acetate are added to the basic medium. Penicillin is added to the stock solution (step a below) to provide a final concentration of 500 U per milliliter. Thallium acetate is added to the basic media (step b below) to provide a final concentration of 1:2000.
Steps
a. Preparation of Stock Solution. To 900ml freshly distilled water, add 50g dextrose and 10g L-arginine HCl. Mix the ingredients at 37°C until dissolved. Bring the final volume up to 1000-ml. Sterilize the solution by filtration using a 0.22-µm filter, dispense into 100-ml aliquots, and store at-70°C until needed.
b. Preparation of Mycoplasma Broth Medium. Add 14.7 g mycoplasma broth base and 0.02g phenol red to 600 ml water, heat to dissolve. Sterilize the solution by autoclaving for 15min at 121°C using a slow exhaust cycle and allow the broth mixture to cool to room temperature. Aseptically add 200ml horse serum, 100 ml yeast extract (15 %), and 100 ml thawed stock solution (step a). Mix the solution completely. Dispense 10-ml aliquots of the broth medium into sterile test tubes and cap. Store broth tubes at 5°C and use within 3 to 4 weeks.
c. Preparation of Mycoplasma Agar Medium. Add 23.8 g mycoplasma agar base to 600ml water. To dissolve, bring solution to a boil and sterilize the solution by autoclaving for 15min at 121°C using a slow exhaust cycle. Place the sterilized medium in a water bath at 50°C Place 200ml horse serum, 100ml yeast extract (15%), and 100ml stock solution (step a) in a water bath at 37°C Allow the components to equilibrate at these respective temperatures. Aseptically add the horse serum, yeast extract, and stock solution to the medium; mix well. Proceed immediately to dispense 10-ml aliquots in 60 × 15-mm petri dishes. Add the fluid as quickly as possible in order to eliminate the problem of the agar solidifying before the medium is dispensed completely. Stack the agar plates into holding racks, wrap in autoclave bags to minimize dehydration, and store at 5°C Use within 3 to 4 weeks.
NOTE This procedure involves a total incubation time of about 35 days for both broth and agar cultures. This schedule is advisable for detecting lower levels of mycoplasma contamination that otherwise might be scored as false negatives.
d. Inoculation of Test Sample. Select a cell culture that is near confluency and has not received a fluid renewal within the last 3 days. Remove and discard all but 3 to 5 ml of the culture medium. Scrape a portion of the cell monolayer into the remaining culture medium using a sterile disposable scraper. For suspension culture systems, take the test sample directly from a heavily concentrated culture that has not received a fresh medium supplement or renewal within the last 3 days. Samples for testing can also be taken directly from thawed ampules that have been stored in the frozen state.
Inoculate 1.0ml of the test cell culture suspension into a mycoplasma broth culture. Inoculate 0.1 ml of the test sample onto an agar culture plate. Incubate the broth culture aerobically at 37°C Observe daily for the development of turbidity and/or shift in pH. Incubate the agar plate anaerobically at 37°C in a humidified atmosphere of 5% CO2-95% nitrogen. After 5 to 7 days of incubation, and again after 10 to 14 days, remove a 0.1-ml sample from the broth culture and inoculate a new agar plate. Incubate these plates anaerobically at 37°C.
Microscopically examine the agar plates weekly for at least 3 weeks for mycoplasma colony formation and growth before considering them to be negative. Observe the plates at 100 to 300 magnification using an inverted microscope.
The positive differentiation of mycoplasma colonies on agar plates, as opposed to air bubbles, tissue culture cells, and pseudocolonies, can be accomplished by subculturing a small section (1 cm2) of the suspicious area of the agar culture into a new broth culture (for examples, see Freshney, 2000).
2. Indirect Method (Staining for DNA)
The bisbenzamide stain concentrate (step a below) should be examined routinely for contamination. Sterilization by filtration diminishes the quality of fluorescence, and fresh stock needs to be prepared periodically. The pH of the mounting medium (step b below) is critical for optimal fluorescence and should also be monitored routinely.
Continuous cells lines, such as ATCC.CCL 81, Vero, African green monkey kidney or ATCC.CCL 96, 3T6 mouse fibroblast, have been used very effectively as indicator cells in the indirect DNA-staining procedure. The use of transformed cell lines is not recommended because they produce large amounts of nuclear background fluorescence, which interfere with the interpretation of the results.
Utilization of the indicator cell with the DNAstaining procedure provides two major advantages. First, the indicator cell line supports the growth of the more fastidious mycoplasma species. Second, both positive and negative control cultures are readily available for direct comparisons with the culture samples being tested.
Steps
a. Preparation of Stain Concentrate. To 100 ml of Hanks' balanced salt solution without sodium bicarbonate or phenol red, add 5.0mg bisbenzamide fluorochrome stain and 10mg thimerosol. Mix thoroughly using a magnetic stirrer for 30 to 45 min at room temperature. The stain is heat and light sensitive. Prepare the concentrate in a brown amber bottle wrapped completely in aluminum foil. Store aliquots at -20°C These are stable for about 1 year.
b. Preparation of Mounting Medium. Combine 22.2ml 0.1M citric acid, 27.8ml 0.2M disodium phosphate, and 50 ml glycerol and adjust pH of mixture to 5.5. Store in a cold room at 5°C.
c. Preparation of Indicator Cell Cultures and Inoculation of Test Samples. Aseptically place a glass coverslip (previously sterilized) into each 60 ×15- mm culture dish. Dispense 3ml Eagle's minimum essential medium with Earle's salts, 100 U/ml penicillin, and 100µg/ml streptomycin plus 10% bovine calf serum into each culture dish. Make certain that each glass coverslip is totally submerged and not floating on top of the medium.
Prepare a single cell suspension of ATCC.CCL 81, the African green monkey kidney cell line Vero, in this medium at a concentration of 1.0 × 105 cells/ml. The 3T6 murine line (ATCC.CCL 96) can be used instead. Inoculate 1 ml of the cell suspension into each culture dish and incubate the cultures overnight in a 5% CO2 95% air incubator at 37°C. Examine the cultures microscopically to verify that the cells have attached to the glass coverslip. Number the top of each culture dish for identification purposes to record the test sample inoculated. Add 0.5 ml of culture medium to each of two cultures for negative controls and 0.2 to 0.5ml of each test sample to each of two culture dishes. Add 0.5 ml ATCC 29052, M. hyorhinis, to each of two cultures for positive controls. Alternatively, a known infected cell line can be used. Return the cultures to the CO2 incubator and allow to incubate undisturbed for 6 days.
d. Fixing, Staining, and Mounting Coverslips. To prepare the staining solution:
- Add 1.0ml of stock concentration (step a) to 100 ml Hanks' balanced salt solution without sodium bicarbonate and phenol red.
- Prepare in a brown amber bottle wrapped in aluminum foil.
- Mix thoroughly for 30 to 45 min at room temperature using a magnetic stirrer.
Remove cultures from the incubator and aspirate the medium from each dish. Add 5 ml of a 1:3 mixture of acetic acid:methanol to each culture dish for 5 min. Do not allow the culture to dry between removal of the culture medium and addition of the fixative. Aspirate each culture dish and repeat the fixation step for 10 min. Aspirate the fixative and let the cultures air dry. Add 5 ml of the staining solution [step d (1-3)] to each culture dish; cover and let stand at room temperature for 30min. Aspirate the stain and rinse each culture three times with 5 ml distilled water.
After the third rinse, aspirate well so that the glass coverslip is completely dry. Let air dry if necessary. Place a drop of mounting fluid (step b) on a clean glass slide. Use forceps to remove the glass coverslip containing the specimen from the culture dish and place face up on the top of the mounting fluid. Add a second drop of mounting fluid onto the top of the specimen coverslip and cover with a larger clean coverslip. Lower both coverslips onto the mounting fluid in such a way as to eliminate trapped air bubbles. Label each slide to identify the specimen being tested and record results.
Observe each specimen under oil immersion, including both the positive and the negative controls, by fluorescence microscopy at 500X. A blue glass excitation filter (BG12 for Zeiss microscopes) is used in combination with a No. 50 barrier filter. Small fluorescing particles indicate mycoplasmal DNA and infection.
Alternative molecular methods readily available in kit form should also be considered. The PCR-based method for mycoplasma detection (Harasawa et al., 1993; Hu and Buck, 1993) in use at the ATCC requires primers based on the DNA sequences in 16S and 23S mycoplasmal rRNA. These amplify DNA from all of the common mycoplasma found in cell cultures to levels easily detected after gel electrophoresis and ethidium bromide staining. Advantages of the method include speed and sensitivity, as well as the ability to detect and identify species of most of the common mycoplasma known to infect cell cultures. Furthermore, it does not suffer from interpretation difficulties . associated with some of the Hoechst or DAPI-stained preparations. Levels of sensitivity compare favorably with the Hoechst stain. Sample sizes need consideration. Detailed methodologies are provided with the kits (see the ATCC website for more details).
C. Protozoa
The overall frequency of infection of cell cultures with protozoans is low but the incidence may be higher if one is working with tissues such as human clinical material and animal tissues such as kidney or colon. The small limax amoebae belonging to the Acanthamoeba (or Hartmanella) genus are ubiquitous in nature and have been isolated from cells and tissues in culture in a significant number of laboratories. Jahnes et al. (1957) first reported spontaneous contamination of monkey kidney cells in culture by such free-living amoebae. The organisms have also been detected as occasional contaminants in such diverse cell lines as dog lymphosarcoma (LS30), HeLa, chick embryo fibroblast-like, and Chang liver cells (Holmgren, 1973). In some cases, protozoans are demonstrably cytopathic in cell culture.
Observation, cytological examination, and attempts at isolation are required in the detection of protozoan contaminants. These techniques are suitable for detecting many of the most common flagellates and amoeboid protozoans, including species of the genera Acanthamoeba, Giardia, Leishmania, Naegleria, and Trypanosoma (for more details, see Hay et al., 1992).
1. Preparation of Solutions and Protozoan Media
Steps
- Trypsin-EDTA: Combine 2.5g trypsin (1:250 Difco certified), 0.3 g EDTA, 0.4 g KCl, 8.0 g NaCl, 1.0 g glucose, 0.58g NaHCO3, and 0.01 g phenol red in 1 liter double-distilled water, sterilize by filtration (0.22- µm Millipore filter), and store at-40°C.
- Hanks" balanced salt solution without divalent cations: Combine 8.0 g NaCl, 0.4 g KCl, 0.05 g Na2HPO4, 0.06 g KH2PO4, and 0.02 g phenol red in 50ml doubledistilled water to dissolve chemicals; then bring volume to 100 ml. Autoclave on slow exhaust for 15 min, adjust pH to 7.2 to 7.4 with sterile 0.4N NaOH, and store at 4°C.
- Giemsa stock solution: For stock solution of stain, combine 40ml glycerol, 65 ml absolute methanol, and 1.0g Giemsa powder. Filter two or three times and store at 4°C.
- Price's buffer (IOX): Combine 6.0g Na2HPO4, 5.0 g KH2P04, and 1.0 liter distilled water. Before use dilute buffer with distilled water to 1X.
- Price's Giemsa stain: Dilute Giemsa stain stock 3:97 with 1X buffer. After staining, discard unused portion.
- ATCC medium No. 400, Diamond's TP-S-1 medium for axenic cultivation of Entamoeba (ATCC medium No. 400): Dissolve one packet of Diamond's TP-S-1 broth base powder in 875 ml distilled water, adjust pH to 7.0 with 0.4N NaOH, and filter through Whatman No. 1 paper. Sterilize at 120°C for 15 min. Aseptically add 100 ml inactivated (56°C for 30min) bovine serum and 25 ml Diamond's TP-S-1 vitamin solution (40X, North American Biologicals), and aseptically dispense 13ml per sterile test tube. Some commercial lots of Diamond's TP-S-1 medium have been shown to be toxic to Entamoeba. To test for toxicity, subculture Entamoeba through three to five passages.
- Locke's solution: Combine 8.0g NaCl, 0.2 g NaCl, 0.2 CaCl2, 0.3 g KH2PO4, 2.5 glucose, and 1.0 liter distilled water and autoclave the solution for 20min at 121°C.
- Diphasic blood agar medium (ATCC medium No. 1011): Infuse 25.0 g beef extract in 250 ml distilled water by bringing to a rapid boil for 2 to 3 min while stirring constantly. Filter through Whatman No. 2 filter paper and add 10.0 g Difco neopeptone, 2.5 g NaC1, and 10.0 g agar. Heat to boiling and filter through Whatman No. 2 paper, make up volume to 500ml with distilled water, and adjust pH to 7.2 to 7.4. Autoclave for 20 min at 121°C, cool mixture to 50°C aseptically add 30% sterile, defibrinated rabbit blood (Editek) to whole mixture, and dispense in sterile tubes and slant. After the slants have set, cover with 3.0ml sterile Locke's solution.
- PYb medium (ATCC medium No. 711): Combine 1.0 g Difco proteose peptone, 1.0g yeast extract, 20.0g agar, and 900.0 ml distilled water. Prepare and sterilize separately each of the following stock solutions and add to the basal medium as indicated to avoid precipitation: CaCl2 (0.05M), 4.0ml; MgSO4.7H20 (0.4M), 2.5 ml; Na2HPO4 (0.25 M), 8.0 ml; and KH2PO2 (0.25 M), 32ml. Make the volume to 1 liter, check that the pH is at 6.5, and sterilize by autoclaving for 25 min at 120°C. Pour into petri dishes and allow to solidify.
- Brain-heart infusion blood agar (ATCC medium No. 807): For the agar component, dissolve 37.0 g Difco BHI broth and 18.0 g agar in 1 liter boiling water. Dispense 5.0ml solution per tube (16 × 125mm) and sterilize for 25 min at 121°C. Cool to 48°C. Add 0.5 ml per tube of sterile, defibrinated rabbit blood and slant. After slants have set, cover with 0.5 ml BHI broth (1.0 liter distilled water and 37.0 g BHI broth) with sterilization by autoclaving at 121°C for 25 min.
- Leishmania medium (ATCC medium No. 811): Combine 1.2g sodium citrate, 1.0g NaCl, and 90.0ml distilled water. Dispense 1.0 ml per tube, autoclave for 25min at 121°C, and cool. Add 1.0ml defibrinated, lysed rabbit blood solution (prepare by mixing equal parts of whole rabbit blood and sterile distilled water and freezing and thawing twice).
- NTYG medium (ATCC medium No. 935): Combine 5.0 g trypticase, 5.0 g yeast extract, 10.0 g glucose, and 1.0 liter distilled water. Dispense 10.0ml per test tube and sterilize. Just before use, add 0.2ml dialyzed, heat-activated bovine serum and 0.1ml defibrinated sheep blood. Protozoan growth media retain stability for at least 3 months if maintained at 4°C with the exception of ATCC medium 400, which maintains stability for 2 to 4 weeks.
2. Preparation of Cell Culture Samples for Inoculation into Protozoan Media
Steps
- Rapidly thaw a frozen ampoule of the sample in a water bath at 37°C.
- Aseptically open the ampoule. Continue to use sterile techniques.
- Transfer 0.8ml of the concentrated cell suspension from the ampoule into a T-25 flask. Save 0.2 ml of the suspension for Giemsa staining (step 3 below).
- Add 7 ml of the appropriate cell culture medium to maintain the culture.
- Incubate at 37°C until the monolayer becomes confluent (3 to 5 days depending on the cell line). Examine the culture microscopically during this incubation period for the presence of (a) movement (i.e., motile cells), (b) intracellular contaminants, and (c) cytopathology.
- Transfer the supernate from the confluent test cell culture to a sterile 15-ml plastic centrifuge tube and retain at room temperature for use in step 12.
- Rinse the cell monolayer (T-25 flask, step 5) with 5ml Ca"- and Mg'- free Hanks' saline and discard saline solution.
- Add 2ml 0.25% trypsin-EDTA solution to the T-25 flask and incubate at 37°C for 10rain.
- Add 7 ml of cell culture medium to the T-25 flask and aspirate gently to obtain a single-cell suspension.
- Dispense aliquots (0.5ml) of the trypsinized single-cell suspension to the following ATCC protozoan growth media: (a) ATCC medium No. 400 (for Entamoeba, Giardia); (b) ATCC medium No. 711 with Enterobacter aerogenes (for Acanthamoeba) [use a wire loop to streak medium No. 711 with E. aerogenes (ATCC 15038) 48h before use]; (c) ATCC medium Nos. 807, 811, and 1011 (for trypanosomatids); and (d) ATCC medium No. 935 (for Naegleria).
- Incubate samples for 7 to 10 days at 35°C and examine microscopically for the presence of flagellate, cyst, and trophozoite forms of protozoa.
- Prepare five wet mounts for each test cell monolayer using the supernate collected in step 6. Examine microscopically with phase contrast for the presence of motile and nonmotile protozoans.
3. Preparation of Culture Cells for Giemsa Staining
Steps
- Aseptically add 1.5 ml of the appropriate culture medium to a sterile Leighton tube containing a coverslip.
- Dispense 0.2ml of the original cell suspension (sample preparation just earlier) into the Leighton tube and incubate at 37°C until the culture is confluent.
- Remove the coverslip from the Leighton tube, fix with absolute methanol for I min, and airdry.
- Stain for 10min with Price's Giemsa, rinse with tap water, and mount the coverslip to a glass slide using Aquamount.
- Examine the slide; use low power (20X) for scanning and high power (100X) for close examination.
Controls
It is recommended that positive controls be included. For example, if cultured cells of the upper respiratory tract are being used, Acanthameba castellanii (ATCC 30010) or Naegleria lovaniensis (ATCC 30569) can be used as positive controls. A. castellanii was isolated from human clinical material. N. lovaniensis strain TS, another nonpathogenic strain of amoebae, was isolated from a Vero cell culture at passage 120. Entamoeba histolytica (ATCC 30042), the common pathogen causing amoebic dysentery, or the nonpathogenic Entamoeba invadens (ATCC 30020) can be used for positive controls if cells are being isolated from the intestinal tract. E. histolytica is a human isolate, and E. invadens strain PZ is a snake isolate.
Comments
The methods described are suitable for the detection of most common protozoan genera (i.e., limax amoebae) that could survive in association with cells in culture. Because cysts and trophozoites closely resemble damaged tissue cells, their presence as occasional contaminants can remain unnoticed. However, cells in cultures infected productively with amoebae of the genus Acanthamoeba frequently become granular and gradually progress to complete disintegration. The time elapsed depends on the inoculum size and whether cysts or the motile trophozoites predominate in the inoculum. The cytopathic effect of amoebic contaminants has been reported, and in some cases the responsible agent has been mistakenly identified as viral in origin. Therefore, frequent observation of the cell culture is particularly stressed when examining for parasitic protozoan contaminants.
The possible presence of other genera (i.e., Entamoeba or trypanosomatids) should be considered not only in experimental studies involving primary tissues, but also with work requiring development or utilization of cell lines. The only known case of an isolation other then an amoeboid protozoan occurred in the isolation of a trypanosomatid from liver tissue. The particular animal and tissue employed provide valuable clues as to the type of protozoan contaminant, the specific media, and staining procedures required.
D. Viruses
Of the various tests applied for detection of adventitious agents associated with cultured cells, those for endogenous and contaminant viruses are the most problematical. Table II lists representative problem viruses. Development of an overt and characteristic cytopathogenic effect (CPE) will certainly provide an early indication of viral contamination; however, the absence of a CPE definitely does not indicate that the culture is virus free. In fact, persistent or latent infections may exist in cell lines and remain undetected until the appropriate immunological, cytological, ultrastructural, and/or biochemical tests are applied. Unfortunately, separate tests are necessary for each class of virus and for specific viruses. Additional host systems or manipulations, e.g., treatment with halogenated nucleosides, may be required for virus activation and isolation (Aaronson et al., 1971). Common screening methods or tests for specific virus classes are listed in Table III.
 |
 |
Without such screens, latent viruses and viruses that do not produce an overt CPE or hemadsorption will escape detection. Some of these could be potentially dangerous for the cell culture technician. For example, Hantaan virus, the causative agent of Korean hemorrhagic fever, replicates in tumor and other cell lines. Outbreaks of the disease in individuals exposed to infected colonies of laboratory rats have been reported separately in five countries. An incident of transmission during passage of a cell line was confirmed in Belgium. As a result of these findings, cell lines expanded in this laboratory were screened using an indirect immunofluorescent antibody assay (LeDuc et al., 1985) and were found to be negative.
Substantial concern over laboratory transmission of the human immunodeficiency viruses is also evident. Cases of probable infection during processing in U.S. laboratories have been described. One, for example, was presumed due to parenteral exposure and another to work with highly concentrated preparations (Weiss et al., 1988). In the latter circumstance, strict adherence to biosafety level 3 containment and practices is essential. A more detailed discussion of safety precautions for work with cell lines in general is provided elsewhere (Caputo, 1988).
ATCC cell lines from selected groups have been screened for HIV-1 using PCR amplification followed by a slot-blot test for envelope and GAG sequences (Ou et al., 1988). The oligonucleotide primer pairs SK 38/39 and SK 68/69 plus SK 19 or SK 70 probes were used. Human cell lines of T-cell, monocytemacrophage, brain and nervous system, B-cell, and gastrointestinal origin plus an array of other primate lines have been examined to date. Only those already known to be infected with HIV-1 have been positive. Additional viruses that could present a substantial health hazard to cell culture technicians include, for example, hepatitis and cytomegaloviruses. Rapid PCR-based tests for these have been described [e.g., Ulrich et al. (1989) and Cassol et al. (1989), respectively]. Other viruses that may present problems generally in cell culture work include ectromelia virus, the causative agent of bovine viral diarrhea (BVDV), and Epstein-Barr virus (EBV). [See also Bolin et al. (1994), Harasawa et al. (1994), and Hay et al. (2000) for testing methodology and further discussion.]
It should be emphasized at the outset that the following protocols represent an expedient compromise established at ATCC to monitor for readily detectable viruses associated with cell lines. Egg inoculations plus select cocultivations and hemadsorption tests were included in addition to routine examinations for CPE using phase-contrast microscopy. Similar general tests are recommended by government agencies in cases where cell lines are to be used for biological production work (Code of Federal Regulations on Animals and Animal Products, 9 CFR 113.34-113.52, revised Jan. 1, 1978; Code of Federal Regulations on Food and Drugs, Subchapter F on Biologics, 21 CFR 630.13 b-c, revised April 1, 1979; IABS, 1989; Lubiniecki and May, 1985). Procedures for reverse transcriptase assays to detect oncogenic viruses are also being applied at the ATCC for selected cell lines.
Because endogenous and most exogenous retroviruses produce no morphological transformation or cytopathology in infected cells, the production of such viruses by cell cultures is generally undetectable except by serological or biochemical means. At ATCC the concentration of particulate material from culture supernates and assay for viral RNA-directed DNA polymerase (RDDP) provide sensitive and reliable means for detecting retrovirus production by cultured cells.
One or more of the following procedures is currently being applied to all cell lines accessioned for the ATCC repository. Tests for specific viruses may be applied through collaborations as described earlier.
1. Examination of Established Cultures for Overt Cytopathogenic Effect or Foci
Steps
- Hold each flask or bottle so that light is transmitted through the monolayer and look for plaques, foci, or areas that lack uniformity. If frozen stocks of cells are to be examined, pool and mix the contents of about 5% of the ampoules from each lot using a syringe with a cannula. Establish cultures for morphological examinations and for tests in the following sections using progeny from such pooled populations.
- Using an inverted microscope equipped with phase-contract optics wherever possible, examine cell culture vessels individually, paying special attention to any uneven areas in gross morphology observed in step 1. Check first using low power. If the cell line is suspect, subculture taking the appropriate safety precautions. Prepare coverslip cultures for further examination. Alternatively, autoclave and discard all suspect cultures. (Stainless-steel collection and sterilizing pans for this purpose can be obtained from the Orem Medical Company.)
- Remove fluid from coverslip cultures that require additional study. Treat with neutral buffered formalin or other suitable fixative. Prepare a wet mount and examine under high power. [Consult Rovozzo and Burke (1973), Hsuing et al. (1994), and Yolken et al. (1999) for examples of cytopathogenic effects and further details.]
2. Application of the Hemadsorption Test
Steps
- Establish test cultures in T-25 flasks using an inoculation density such that the monolayers become confluent in 48 to 72 h.
- Prepare washed red blood cell suspensions on the day the test is to be performed. Pack the erythrocytes from 5ml of the purchased suspensions by centrifugation at 100g and resuspend in 35 ml Hanks' saline without divalent canons. Repeat three times and resuspend the final pellet to yield a 0.5% suspension (v/v) of red blood cells in saline.
- Remove the culture fluid and rinse the test monolayers with 5 ml Hanks' saline minus divalent cations.
- Add 0.5ml each of the suspensions of chick, guinea pig, and human type O erythrocytes from step 2. Then place the flask with monolayer down at 4°C for 20 rain.
- Observe macroscopically and microscopically under low power for clumping and adsorption of red blood cells to the monolayer.
- Repeat steps 2-4 on all test cultures not exhibiting hemadsorption before recording a negative result. [A suitable positive control can be established by infecting a flask of rhesus monkey kidney cells with 0.2 ml of undiluted ATCC VR-95 (influenza virus strain A/PR/8/34) 48 to 72h before testing.]
3. Egg Preparation
Steps
- Drill a small hole in the egg air sac (blunt end) using the electric drill (Cole-Parmer) and a 1/16-in. burr-type bit or an 18-gauge needle in this and subsequent operations; work with sterile instruments. Swab areas of the shell to be drilled with 70% ethanol before and after each manipulation. The drill bits may be placed in 70% ethanol before use.
- Using the candling lamp (Nasco), locate the area of obvious blood vessel development and, at a central point, carefully drill through the shell, leaving the shell membrane intact.
- Place 2 or 3 drops of Hanks' saline on the side hole and carefully pick through the shell membrane with a 26-gauge syringe needle. The saline will seep in and over the chorioallantoic membrane (CAM) to facilitate its separation from the shell membrane.
- Apply gentle suction to the hole in the air sac using a short piece of rubber tubing with one end to the mouth and the other pressed to the blunt end of the egg. Use the candling lamp to monitor formation of the artificial air sac over the CAM.
- Seal both holes with squares of adhesive or laboratory tape and incubate the eggs horizontally at 37°C. Standard cell culture incubators and walk-in rooms are entirely adequate for egg incubations. Highhumidity or air/CO2 boxes are not satisfactory.
4. Egg Inoculations
Steps
- Obtain suspensions of test cells in the appropriate growth medium and adjust the concentration such that 0.2 ml contains 0.5 to 1 × 107 cells.
- Remove the seal from side holes in the embryonated eggs and inject 0.2ml of the cell suspension onto the CAM of each of 5 to 10 eggs.
- Using the candling lamp, examine the embryos 1 day after adding the cell suspension; discard any embryos that have died. Repeat the examination periodically for 8 to 9 days.
- If embryos appear to be viable at the end of the incubation period, open the eggs over the artificial air sac and examine the CAM carefully for edema, foci, or pox. Check the embryo itself for any gross abnormalities such as body contortions or stunting.
- In cases in which viral contamination is indicated, repeat steps 1-4 both with a second aliquot of the suspect cells and with fresh fluid samples from eggs in which the embryos have died or appear abnormal. Positive controls may be established by inoculating eggs with influenza virus, Newcastle disease virus, and/or Rous sarcoma virus.
5. Cocultivation Trials
Steps
- Select two appropriate cell lines for cocultivation with each cell line to be tested. The lines chosen will depend on the species from which the test cell line originated. For example, for a human cell line, one could cocultivate with ATCC CCL 75 (WI-38), ATCC CCL 171 (MRC-5), or primary human embryonic kidney (HEK) cells. A cell line from a second species of choice in this example could be ATCC CCL 81 (Vero) originating from the African green monkey.
- Inoculate a T-75 flask with 106 cells from each line in a total of 8 ml of an appropriate growth medium. In some cases, the inocula may have to be adjusted in an attempt to maintain both cell populations during the cocultivation period. For example, if a very rapidly proliferating line is cocultivated with a test line that multiplies slowly, the initial ratio of the former to the latter could be adjusted to 1:10. Similarly, the population that multiplies slowly might have to be reintroduced to the cocultivation flasks if it were being overgrown by the more rapidly dividing cells.
- Change the culture fluid twice weekly and subcultivate the population as usual soon after it reaches confluence.
- Examine periodically for CPE and hemadsorption over a 2- to 3-week period at minimum, using procedures described earlier.
Viral isolates may be identified through standard neutralization (hemadsorption inhibition, plaque inhibition, hemagglutination inhibition) or complement fixation tests. The ATCC virology department retains and distributes antisera to many viral serotypes, and identification can be accomplished readily.
6. Reverse Transcriptase Assays
Positive serological assays for retrovirus antigens in cells and cell packs indicate that a retrovirus genome is present, but these assays do not indicate whether release of progeny virus particles is occurring. It has been found that the concentration of particulate material from culture supernates and the assay for viral RDDP (Baltimore, 1970; Temin and Mizutani, 1970) provide a sensitive and reliable means for detecting retrovirus production by cultured cells.
a. Preparation of Cell Cultures. Cell cultures to be examined for the production of retrovirus should be cultured by the methods and in the media that are optimal for the particular cells. It is important that the cells be in good condition and not undergoing degeneration and autolysis.
Steps
- When adherent cell cultures are about 50 to 60% confluent, or when suspension cultures are at a cell density about 50% of the maximum, completely replace the medium and reincubate the cultures.
- Harvest fluid approximately 24 h after feeding.
b. Processing of Culture Fluid
Steps
- Collect culture medium aseptically.
- Clarify medium by centrifugation at 1000 to 3000g for 10rain at 4°C. Decant and save the clarified supernates and discard sedimented materials.
- The clarified medium contains 0.15M NaCl; add 5.0M NaCl to a final concentration of 0.5 M NaCl. Calculate the volume of 5.0M NaCl according to the formula 0.15(V1) + 5.0(V2) = 0.5 (1/1 + V2), where 1/1 is the volume of clarified culture fluid and V2 is the volume of 5.0M NaCl to be added. Mix well. If the medium becomes cloudy after adding NaCl, centrifuge at 10,000 g for 10rain and save the supernate.
- To 2 volumes of clarified supernate containing 0.5M NaCl, add 1 volume 30% PEG 6000 in 0.5M NaCl. Mix well.
- Allow precipitation to occur for at least 1 h while holding in wet ice. At this point samples may be held overnight at 4°C if necessary.
- Centrifuge at 7000g for 10 min.
- Decant and discard the supernates.
- Drain the pellets thoroughly while holding at 4°C.
- Resuspend the pellets in 50% (v/v) buffer A [0.05 M Tris-HCl, pH 7.5, 0.1M KCl, 0.5mM EDTA, 10mM dithiothreitol, 0.05% (v/v) Triton X-100, and 50% glycerol]. Care must be taken to ensure that pellets are completely resuspended.
- Store resuspended pellets at-20°C Aliquots are used for RDDP assays.
c. Assay of RNA-Directed DNA Polymerase Activity. This procedure is based on that of Gallagher and Gallo (1975).
Solutions
- Stock mix: 0.5% (v/v) Triton X-100, 1.13M KCI.
- Template-primer solutions (P-L Biochemicals). Mix A: Combine 1 mg/ml poly(rA)-poly(dT)12-18s with 0.01 M Tris-HCl, pH 7.5, and 0.1M NaCl. Mix B: Combine 1 mg/ml poly(dA)-poly(dT)12-18 with 0.1M NaCl and 0.01 M Tris-HCI, pH 7.5.
- Working mixtures of template-primer solutions: Mix stock mix and template-primer solutions in 3:2 (v/v) ratio.
- Reaction cocktail: Evaporate 250 µl [3H]TTP (carrier-free, New England Nuclear 221-X) to dryness under vacuum. Redissolve in 720/µl H2O before adding the following components (volumes given are for 10 tubes): 1.0M Tris-HCl, pH 7.8 (40µl), 0.2M dithiothreitol (40µl), 0.01 M MnCI2 (50µl). Add MnCI2 last (just before initiating reactions).
Steps
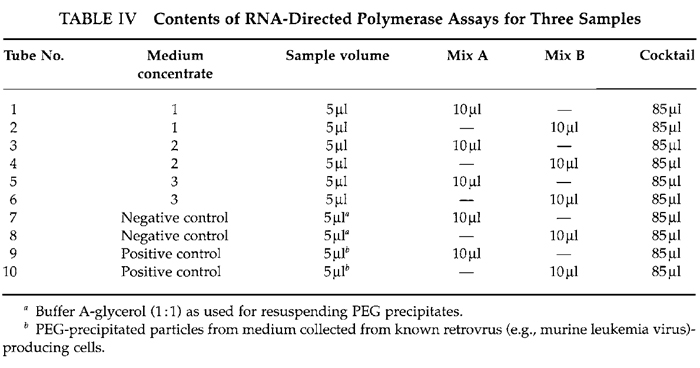
- Distribute culture medium concentrates and positive and negative control samples into siliconized 10 × 75-mm assay tubes. (a) For positive controls, use concentrates prepared from culture media of cell cultures known to be producing retroviruses. (b) For negative controls, use buffer A-glycerol (Table IV).
- Add appropriate template-primer mix to each tube and mix with a vortex mixer. Hold tubes in wet ice for 15 min.
- Initiate reactions by adding reaction cocktail to each tube and mixing. Allow reactions to proceed for 30min in a 37°C water bath.
- At the end of incubation period, remove tubes to an ice bath; terminate reactions by adding 25B1 0.1M EDTA per tube (Sethi and Sethi, 1975).
- Spot 100µl from each tube onto appropriately numbered DE-81 filters. Allow liquid to soak into filters.
- Wash batches of filters with gentle manual swirling in at least 10ml (per filter) of 5% (w/v) Na2HPO4 . 7H20. Repeat for a total of six washes (Sethi and Sethi, 1975).
- Wash twice with distilled H20 and twice with 95% ethanol and arrange filters on cardboard covered with absorbent paper. Dry thoroughly under a heat lamp.
- Place each filter in a separate numbered scintillation vial, add 10 ml PPO-POPOP scintillation cocktail (Betafluor) to the vial, and count in a liquid scintillation counter (Beckman LS-3133) using a tritium window.
 |
Comments
A number of precautions must be observed in the interpretation of the results obtained in this assay. If the cultures to be tested are very heavy and undergoing autolysis, a large amount of cellular DNA-directed DNA polymerase may be associated with microsomal particles in the culture medium. These particles are concentrated by the polyethylene glycol procedure just as virus particles are. Because cellular DNA polymerases do not exhibit an absolute specificity for a DNA template, a certain level of [3H]TMP ([3H]thymidylate) incorporation directed by an RNA template will result from cellular polymerase activity. If a high level of DNA-directed cellular polymerase activity is present in medium concentrates, the (sometimes high) degree of incorporation by these enzymes can mask true RDDP activity, which may be present. Consequently, it is important that media be collected from healthy, actively growing cultures.
It must be remembered that enzymes that catalyze the polymerization or terminal addition of [3H]TMP with a poly(rA) 9 oligo(dT) template-primer are not exclusively viral (Harrison et al., 1976). poly(rA) DD-CC oligo(dT) is generally employed because the activity of retroviral RDDP is usually greater with that template-primer than it is when measured by the incorporation of dGMP (deoxyguanylate) directed by poly(rC) 9 oligo(dG1) or by methylated derivatives of the poly(rC) template; however, DNA synthesis directed by poly(rC) 9 oligo(dG) is more specific for viral enzyme. Consequently, medium concentrates that show incorporation of [3H]TMP with the poly(rA) template should be tested for the incorporation of [3H]dGMP directed by a poly(rC) template.
Incorporation of isotopic precursors into macromolecular form is generally detected by the precipitation of macromolecules with trichloroacetic acid after the enzymatic reaction is terminated. Although background levels of radioactivity may be somewhat higher by the use of adsorption to and elution from ion-exchange filter paper, the ion-exchange procedure obviates the need for a filtration manifold, which is generally employed for acid precipitation. Also, the batch method employed allows many more samples to be processed efficiently.
7. A Rapid PCR-Based Procedure for Detecting the Presence of HIV and HTLV RNA in ATCC Cell Lines
An additional high-throughput method has been adopted to screen for HIV and HTLV RNA in ATCC cell lines using quantitative PCR. Priorities include lines, which might be expected to support growth of these viruses, namely T cells, macrophages and monocytes, lines from the brain and nervous system, lines of gastrointestinal origin, selected human hybridomas, and others.
Steps
- Total RNA is extracted from 106 cells of selected cell lines (Qiagen, Valencia, CA).
- After quantitative and qualitative analyses using the Bioanalyser 2100, 500ng of RNA is reverse transcribed in the presence of a mixture of random hexamers and oligo(dT). For real-time PCR (ABI PRISM 700, sequence detector, Foster City, CA), 12.5ng of initial RNA (0.5 µl of cDNA reaction) is used.
- Specific primers and TaqMan probes are used individually to measure levels of HIV-1, HTLV-1, and GAPDH transcripts. For HIV-1 and HTLV-1, the Sybr Green I dye assay is also used.
Notes and Results
Both the SybrGreenI dye (Molecular Probes, Eugene, OR) and TaqMan assays for HIV-1 and HTLV- 1 show an amplification signal. GAPDH transcripts, used as an internal control, are detectable in all the cell lines tested. Every sample is run on triplicates, and an average is calculated. Absolute quantities of HIV, HTLV, and GADPH are calculated using the standard DNA method as described in User Bulletin 2 (PE Applied Biosystems, Foster City, CA).
We have used two different detection methods: Sybr Green I dye or the TaqMan probe method. Sybr Green I is a double-stranded DNA-binding dye, which, when added into the PCR mix, binds to the amplicon, the double-stranded DNA fragment produced during PCR. As the PCR progresses, more amplicons are created. Due to the binding of Sybr Green I dye to all double-stranded DNA, the increase in fluorescence is proportionate to the amount of PCR product. In addition to forward and reverse primers, TaqMan technology uses a sequence-specific oligonucleotide labeled with fluorescent dye at the 5' end and a quencher dye at the 3' end. While the probe is intact, the proximity of the quencher dye reduces the fluorescence emitted by the reporter dye by fluorescence resonance energy transfer (FRET) through space. If the target sequence is present, the probe anneals downstream from one of the primer sites and is cleaved by the 5' nuclease activity of Taq DNA polymerase during primer extension (Figs. 1A and 1B). The cleavage of the probe separates the reporter dye from the quencher dye, increasing the reporter dye signal (Fig. 1C). Additional cleavage of the probe occurs at every cycle, resulting in an increase of the fluorescent signal proportional to the amount of the amplicon. Thus the presence of the TaqMan probe enables detection of the specific amplicon as it accumulates during PCR cycles.
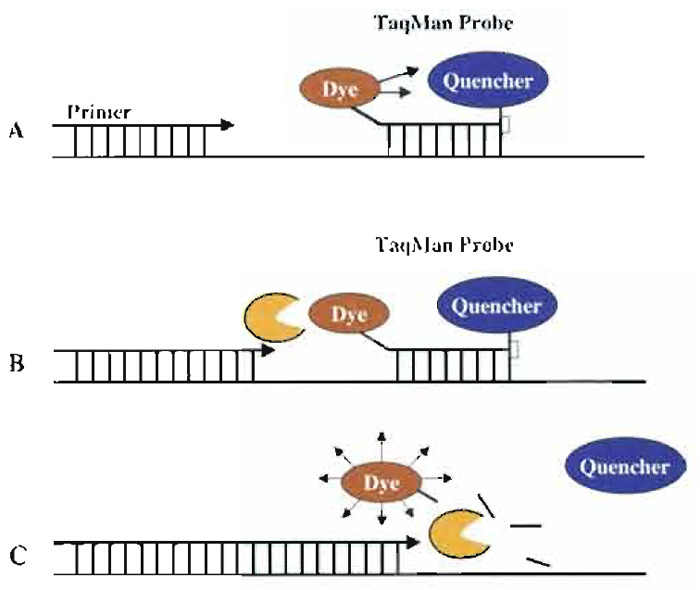 |
| FIGURE 1 Description of 5' nuclease assay. (A) Sequence-specific primers and dual-fluorescence TaqMan probe anneal to complementary sequences in the DNA template. Due to frequency resonance energy of transfer emission of the fluorescence dye (reporter) is reduced significantly by the presence of the proximal quencher. (B) Due to Taq polymerase activity, primer extension and synthesis of a complementary strand occur. (C) While extending, due to its 5' nuclease activity, the Taq polymerase cleaves the TaqMan probe and enables the release of a fluorescent signal by the reporter. |
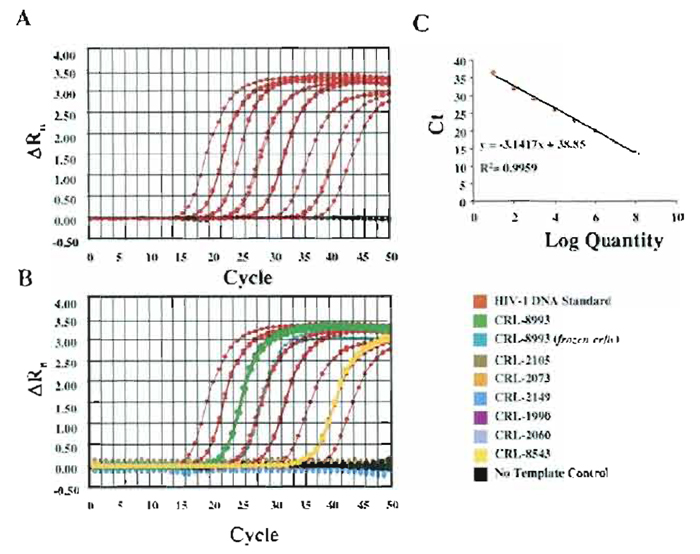 |
| FIGURE 2 Real-time PCR assays for the detection and quantitation of HIV. Amplification plots (A) and standard curves (B) generated for the quantitation of HIV. (C) Amplification plots of unknown templates (total RNA from human cell lines). |
HIV- and HTLV-specific primers and probes were designed to avoid false-negative samples due to nucleotide variation in the target sequence (Desire 2001; Schutten, 2000; Bisset, 2001). Blast search of GenBank indicated that the probe and primer set used in these analyses will detect the majority of HIV-1 subtypes and HTLV-I. Total RNA was extracted from frozen cell pellets for all the cell lines used in the test. For CRL-8993, total RNA was used from both frozen and fresh cells as a comparison. During RNA extraction, DNase I was added to avoid contamination of RNA extracts with genomic DNA. Extracted RNA was evaluated using the 2100 Bioanalyzer (Agilent Technologies, Wilmington, DE). Although the amount of total RNA extracted from frozen cells is significantly lower than the total RNA extracted from fresh cells, the RNA extracted from frozen cells is sufficient for several RT-PCR assays and no RNA degradation was observed. Total RNA (300ng) was reverse transcribed in a 20-µ reaction using Superscript (Invitrogen, Carlsbad, CA) and 1 µl of the cDNA product was used for real-time PCR. We have used both Sybr Green and TaqMan methods to quantitate and detect HIV-1 and HTLV-I viruses in human cell lines. For absolute quantitation, known amounts of plasmid DNA containing full-length or partial sequences of HIV and HTLV were used in parallel with the unknown templates. PCR was performed using the same set of primers and the TaqMan probe for both standard and sample cDNA. Amplification plots and standard curves were generated for HIV (Fig. 2A) and HTLV (data not shown). The absolute amounts of HIV and HTLV for each sample were calculated based on the standard curves (Figs. 2B and 2C). We have also analyzed levels of GAPDH as an endogenous control, and standard curves for GAPDH were generated using plasmid DNA containing a GAPDH cDNA (Fig. 3). Absolute amounts of GAPDH were also calculated based on the standard curve method.
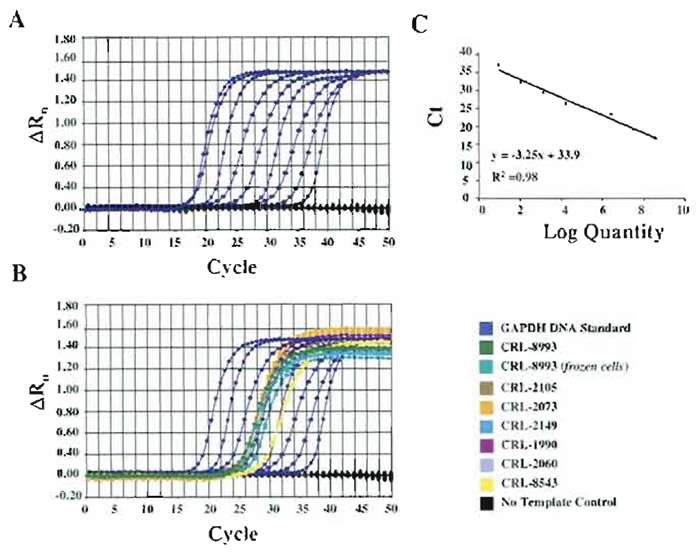 |
| FIGURE 3 Real-time PCR assays for the quantitation of endogenous GAPDH transcripts as an internal control. Amplification plots (A) and standard curves (B) generated for the quantitation of GAPDH. (C) Amplification plots of endogenous GAPDH from each specific cell line. |
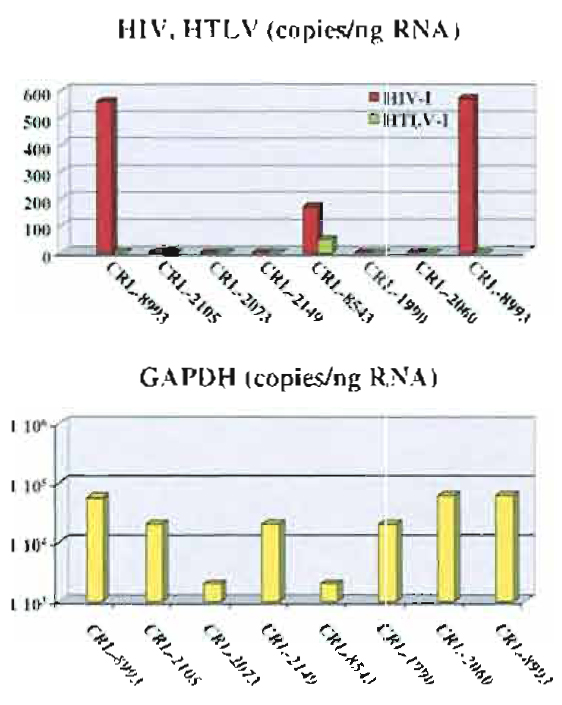 |
| FIGURE 4 Detection and quantitation of HIV, HTLV, and GAPDH transcripts based on real-time PCR analyses. |
Quantitation of copies of viral HIV and HTLV, as well as quantitation of endogenous GAPDH, is shown in Table V and Fig. 4. Sybr Green assays were run for the quantitation of HIV, HTLV, and GAPDH, analyses were performed using the standard curve method, and similar results were obtained (data not shown). Therefore, if primers are well designed and PCR conditions are such that do not generate nonspecific amplicons or primer dimers, testing for the presence of viral RNA using Sybr Green I dye could be less expensive and as effective as the TaqMan assay. As shown in Figs. 3 and 4, HIV and HTLV viral RNA was detected only in CRL- 8993 and CRL-8543 cell lines. CRL-8993 is a lymphoid cell line reportedly positive for HIV, and CRL-8543 is also reported to contain HIV and HTLV viruses. CRL-8993 was used as a control, and total RNA was extracted from both fresh culture and frozen cells. As shown in Fig. 4, the difference in absolute copies of either GAPDH or HIV transcripts between the two RNA extracts is insignificant, suggesting that total RNA extracted from frozen cells could be used effectively for RT-PCR analyses.
 |
In summary, we have established a quick and accurate method for the detection of HIV-1 and HTLV-I viruses. Given the high sensitivity of real-time PCR, very low amounts of total RNA are needed for PCR analyses. Therefore, RNA can be extracted directly from frozen cell pellets and particular cell lines can be tested for the presence of viral RNA prior to cultivation. Real-time PCR offers the possibility of detecting and quantitating as low as 10 copies of viral DNA.
IV. GENERAL COMMENTS
The microbial contamination and viral infection of cell lines are still extremely serious problems. Mycoplasmal infection has been especially well studied, and the incidence of problems has been documented through government-funded programs. Screening results reported within the past two decades showed that as many as 4 to 33% of cultures tested were infected with one or more species of mycoplasma (Hay et al., 1989). It is absolutely imperative that cell lines used in research or production work be tested routinely for such adventitious infection. The comparative cost in time and materials is extremely small. Rewards in terms of research or production reliability are substantial.
Testing for viral infection is more problematical in that it is expensive and multiple tests are required to provide even a limited degree of assurance on freedom from infection. We recommend consideration of screening on a case-by-case basis depending on anticipated use for the line, funding available, and a riskversus- benefit analysis. Of course, a potential health hazard for cell culture technicians is a major concern.
Finally, it is also critically important to verify the identity of cell lines employed. Hukku et al. (1984) documented the incidence of cross-contamination of cell lines reporting misidentifications in excess of 35%.
Thus, a reasonably rigorous authentication pro-gram must include not only reliable tests to ensure an absence of microbial infections (including mycoplasma), but also cell species verification. Methods are detailed elsewhere (Hay et al., 2000, 1992) as are precautions required to avoid operator-induced contamination during routine processing (Hay, 1991).
References
Aaronson, S. A., Todaro, G. J., and Scolnick, E. M. (1971). Induction of murine C-type viruses from clonal lines of virus-free BALB/3T3 cells. Science 174, 157-159.
Baltimore, D. (1970). RNA-dependent DNA polymerase in virions of RNA tumor viruses. Nature (Lond.) 226, 1209-1211.
Bisset L. R. (2001). Quantification of in vitro retroviral replication using a one-tube real time RT-PCR system incorporating direct RNA preparation. J. Virol. Methods. 91(2), 149-155.
Bolin, S. R., Ridpath, I. E, Black, J., Macy, M., and Roblin, R. (1994). Survey of cell lines in the American Type Culture Collection for bovine viral diarrhea virus. J. Virol. Methods 48, 211-221. Caputo, J. (1988). Biosafety procedures in cell cultures. J. Tissue Cult Methods 11, 223-228.
Cassol, S. A., Poon, M.-C., Pal, R., Naylor, M. J., Culver-lames, J., Bowen, T. J., Russel, J. A., Krawetz, S. A., Pon, R. T., and Hoar, I. (1989). Primer mediated enzymatic amplification of cytomegalovirus (CMV) DNA. J. Clin. Invest. 83, 1109-1115.
Chen, T. R. (1977). In situ demonstration of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Exp. Cell Res. 104, 255-262.
Desire, N. (2001). Quantification of HIV type 1 proviral load by TaqMan real time PCR. J. Clin. Microbiol. 39(4), 1303-1310.
Fogh, J. (1973). Contaminants demonstrated by microscopy of living tissue cultures or of fixed and stained tissue culture preparations.In "Contamination in Tissue Culture" (J. Fogh, ed.), pp. 65-106. Academic Press, New York.
Freshney, R. I. (2000). "Culture of Animal Cells: A Manual of Basic Technique," 4th Ed., Wiley-Liss, New York.
Gallagher, R. E., and Gallo, R. C. (1975). Type C RNA tumor virus isolated from cultured human acute myelogenous leukemia cells. Science 187, 350-353.
Harasawa, R., Kazumasa, H., Tanabe, H., Takada, Y., and Mizusawa, H. (1994). Detection of adventitious pestivirus in cell cultures by polymerase chain reaction using nested-pair primers. Jpn. Tissue Cult. Assoc. J. 12, 215-220.
Harrison, T. A., Barr, R. D., McCaffrey, R. P., Sarna, G., Silverstone, A. E, Perry, S., and Baltimore, D. (1976). Terminal deoxynucleotidyl transferase in AKR leukemia cells and lack of relation of enzyme activity to cell cycle phase. Biochem. Biophys. Res. Commun. 69, 63-67.
Hay, R. J. (1991). Operator-induced contamination in cell cultures systems. Aeres-Sorono Symp. Dev. Biol. Stand. 75, 193-204.
Hay, R. J., Caputo, J., and Macy, M. (1992). "ATCC Quality Control Methods for Cell Lines," 2nd Ed. ATCC, Rockville, MD.
Hay, R. J., Cleland, M. M., Durkin, S., and Reid, Y. A. (2000). Cell Line Preservation and Authentication in "Animal Cell Culture" (J. R. W. Masters, ed.) Oxford Univ. Press, New York.
Hay, R. J., Macy, M. L., and Chen, T. R. (1989). Mycoplasma infection of cultured cells. Nature 339, 487-488.
Holmgren, N. B. (1973). Contamination in tissue culture by parasites. In "Contamination in Tissue Culture" (J. Fogh, ed.), pp. 195-203. Academic Press, New York.
Hsiung, G. D., Fong, C. K. Y., and Landry, M. L. (1994). "Hsuing's Diagnostic Virology," 4th Ed. Yale Univ. Press, London.
Hu, M., and Buck, C. (1993). Application of polymerase chain reaction technique for detection of mycoplasma contamination. J. Tissue Cult. Methods 15, 155-160.
Hukku, B., Halton, D. M., Mally, M., and Peterson, W. D., Jr. (1984). Cell characterization by use of multiple genetic markers in eukaryotic cell cultures. In "Eukaryotic Cell Cultures, Basics and Applications" (R. T. Acton and J. D. Lyn, eds.), pp. 13-31. Plenum Press, New York.
IABS (1989). "Continuous Cell Lines as Substrates for Biologicals." IABS Symposium on Developments in Biological Standardization, Vol. 70. Karger, Basel.
Jahnes, W. G., Fullmer, H. M., and Li, C. P. (1957). Free-living amoebae as contaminants in monkey kidney tissue culture. Proc. Soc. Exp. Biol. Med. 96, 484-488.
LeDuc, J. W., Smith, G. A., Macy, M. L., and Hay, R. J. (1985). Certified cell lines of rat origin appear free of infection with hantavirus. J. Infect. Dis. 152, 1081-1082.
Lubiniecki, A. S., and May, L. H. (1985). Cell bank characterization for recombinant DNA mammalian cell lines. Dev. Biol. Stand. 60, 141-146.
McGarrity, G. J. (1982). Detection of mycoplasmal infection of cell cultures. Adv. Cell Cult. 2, 99-131.
Ou, C.-Y., Kwok, S., Mitchell, S. W., Mack, D. H., Sninsky, J. J., Krebs, J. W., Feorino, P., Warfield, D., and Schochetman, G. (1988). DNA amplification for direct detection of HIV-1 in DNA of peripheral blood mononuclear cells. Science 239, 295-297.
Rovozzo, G. C., and Burke, C. N. (1973). "A Manual of Basic Virological Techniques." Prentice-Hall, Englewood Cliff's, NJ.
Schutten, T. (2000). Development of real-time Q RT-PCR for detection of HIV-2 RNA in plasma. J. Virol. Methods 88, 81-87.
Sethi, V. S., and Sethi, M. L. (1975). Inhibition of reverse transcriptase activity of RNA tumor viruses by fagaronine. Biochem. Biophys. Res. Commun. 63, 1070-1076.
Temin, H. M., and Mizutani, S. (1970). RNA-dependent DNA polymerase in vMons of ROUS sarcoma virus. Nature (Lond.) 226, 1211-1213.
Ulrich, P. P., Bhat, R. A., Seto, B., Mack, D., Sninsky, J., and Yvas, G. N. (1989). Enzymatic amplification of hepatitis B virus DNA in serum compared with infectivity testing in chimpanzees. J. Infect. Dis. 160, 37-43.
Uphoff, C. C., and Drexler, H. G. (2001). Prevention of mycoplasma contamination in leukemia-lymphoma cell lines. Hum. Cell 14, 244-247.
Weiss, S. H., Goedert, J. J., Gartner, S., Popovic, M., Waters, D., Markham, P., Veronese, E M., Gail, M. H., Barkley, W. E, Gibbons, J., Gill, E A., Leuther, M., Shaw, G. M., Gallo, R. C., and Blattner, W. A. (1988). Risk of human immunodeficiency virus (HIV-1) infection among laboratory workers. Science 239, 68-71.
Yolken, R. H., Lennette, D. A., Smith, T. E, and Warner, J. L. (1999). "Manual of Clinical Microbiology," 7th Ed. ASM Press, Washington, DC.
Support our developers




