Detection of Protein-Protein Interactions in vivo Using Cyan and Yellow Fluorescent Proteins
I. INTRODUCTIONThe phenomenon of fluorescence resonance energy transfer (FRET) describes the transfer of energy from one fluorophore to another through dipole-dipole interaction (Forster, 1946, 1948). It is a sensitive "molecular ruler" that can detect molecular associations within a range of 100Å (Stryer, 1978). Early work using FRET to measure biological associations relied on either fluorescent analogs of biomolecules or fluorescent antibodies as donors and acceptors. However, the utility of this technique in detecting biological associations was limited by the availability of monoclonal antibodies that recognize epitopes of the receptor where maximum energy transfer can occur without disrupting the interactions under examination. This limitation was overcome with the use of spectral variants of the green fluorescent protein (GFP) (Tsien, 1998). The use of GFP variants such as CFP (cyan) and YFP (yellow) in FRET analysis has the additional advantage of allowing detection of intracellular associations in living cells. While most of the applications of FRET have employed fluorescence microscopic imaging methods, it has recently become feasible to perform FRET analysis using flow cytometry. For example, it has been used successfully to determine the ligand-independent association between different Fas receptors (Siegel et al., 2000). More recently, flow cytometric FRET analysis has been used to study the interaction between histone acetylase PCAF and histone deacetylase HDAC1 and other chromatinbinding proteins (Kanno et al., 2004; Yamagoe et al., 2003), to detect the presence of Bacillus anthracis spores in a test biological sample (Zahavy et al., 2003) and to detect caspase activation during apoptosis induction (He et al., 2003). Flow cytometry-based FRET analysis permits the screening of a large number of interactions within a short time and will be a useful technique in the screening of molecular associations in the proteomics era. We will focus our discussion to flow cytometry-based FRET analysis. For a more detailed description of fluorescence microscopic FRET analysis, readers are referred to the following link: http://www. stke. org/cgi / content / full / OC_sitrans;2000 / 38 / pl1.
II. MATERIALS AND INSTRUMENTATION
The FACS Vantage SE flow cytometer is from BD Bioscience (San Jose, CA). RPMI 1640 without phenol red (Cat. No. 11835055), penicillin and streptomycin (Cat. No. 10378016), L-glutamine (Cat. No. 25030081), phosphate-bultered saline (PBS, Cat. No. 14190250), and trypsin versene (Cat. No. 15040066) are from Invitrogen (Carlsbad, CA). Fetal calf serum (FCS) is from Biofluids (Cat. No. 200P-500). HEK 293T cells are from ATCC (Cat. No. CRL-11268). Fugene 6 (Cat. No. 1814443) and propidium iodide (Cat. No. 1348639) are from Roche. The CFP (Cat. No. 6900-1) and YFP (Cat. No. 6006-1) parental plasmids are from BD Clontech (CA). Flowjo software is from Treestar Inc. (San Carlos, CA). β-Mercaptoethanol (Cat. No. M-6250) and bovine serum albumin (BSA, Cat. No. A-7906) are from Sigma (MO).
III. PROCEDURES
A. Transfection of FRET Constructs
- Split HEK 293T cells by washing the cell monolayer with 10ml PBS and incubating cells at 37°C for 5-10min in 1× trypsin versene (2-5 ml). Seed cells at 2.5 × 105 cells per well in 1 ml volume of phenol red-free RPMI 1640 (supplemented with 10% FCS, 100 units/ml of penicillin and streptomycin, 2 mM of L-glutamine, and 54µM β-mercaptoethanol) in a 12- well culture plate.
- Incubate cells in a CO2 incubator for 16-20h. Cells should be 70-90% confluent at the time of transfection.
- Prepare the DNA mixture by mixing an equal molar ratio of CFP to YFP plasmids in an Eppendorf tube. The optimal molar ratio of CFP to YFP plasmids may be different for different FRET pairs and should be determined empirically. The total amount of DNA should be 1 µg per 3 µl of Fugene 6 for transfection in 12-well plate.
- Add 100µl of serum-free Dulbecco's Modified Eagles Medium to an Eppendorf tube. Add 3µl of Fugene 6 to the serum-free medium. Gently tap the tube to mix it. Incubate at room temperature for 5 min.
- Add the diluted Fugene 6 solution from step 4 to the DNA mixture in step 3. Gently tap the tube to mix well. Incubate the mixture at room temperature for 15 min.
- Add the DNA/Fugene 6 mixture from step 5 to the cells dropwise. Gently swirl the plate to distribute the mixture evenly.
- Incubate the cells in a 37°C CO2 incubator for 24-48 h. It is not necessary to change the medium after transfection.
- After 24-48h, aspirate the medium from the wells. Resuspend the cells in 1 ml PBS supplemented with 2% BSA. Spin the cells down at 1500rpm in a Beckman tabletop centrifuge for 5 min at 4°C.
- Decant the supernatant. Resuspend cells in 1 ml of PBS supplemented with 2% BSA. Keep cells on ice until they are ready for analysis. Alternatively, cells can be fixed in 4% paraformaldehyde until they are ready for analysis.
B. Collection of Data on FACS Vantage SE
1. Setup of Flow Cytometer
Analyze cells on a FACS Vantage SE flow cytometer (equipped with an ILT air-cooled argon laser and a Spectra Physics Model 2060 krypton laser). Tune the argon to 514nm for direct excitation of YFP and the krypton laser to 413 nm for excitation of CFP. Replace the forward and side scatter filters with the 513/10 bandpass filter. Use a filter with 470/20-nm bandpass for CFP detection in the P6 channel and filters with 546/10-nm bandpass for YFP and FRET detection in P3 and P5 channels, respectively. Direct YFP fluorescence for detection in the P7 channel for interlaser compensation (see step 2). Use a 505LP dichroic mirror to separate the CFP and FRET signals in P5 and P6 channels. Adjust the fluidics pressure to 29-30psi.
2. Compensations
Perform electronic compensation to remove CFP emission from the FRET channel (P5-P6). Perform interlaser compensation between CFP and YFP using Omnicomp circuitry to remove YFP excitation from the 413-nm laser. Alternatively, compensations can be performed "off-line" using softwares such as Flowjo during data analysis.
3. Run and Collect Samples
Collect 50,000 live events per sample. To facilitate distinction of live and dead cells, 1µg/ml of propidium iodide can be added to cells prior to collection of events on the flow cytometer.
4. Analysis of Data
Analyze collected data using Flowjo software. Control samples transfected with noninteracting FRET pairs should be used as negative controls to determine the baseline FRET signal. An example of step-bystep analysis of the homotypic interaction of the ectodomain of p60 TNFR-1 using flow cytometric FRET analysis is given in Fig. 1. TNFR-1 forms ligandindependent complexes through a domain at the N terminus of the receptor called the preligand assembly domain (PLAD) (Chan et al., 2000). The cytoplasmic tail of TNFR-1 is replaced by CFP or YFP. The resulting fusions are expressed in HEK 293T cells. A positive interaction between the differentially tagged TNFR-1 ectodomains is observed as the FRET signal increases over cells expressing only the CFP-tagged receptor (Fig. 1a). Moreover, when TNF is added to the cells, a ligand-dependent rearrangement of the complex results in further increase of the FRET signal (Fig. 1a). The interaction between different TNFR-1 chains is specific, as replacing the fluorescence donor or acceptor with another TNFR-like receptor DR4 abolishes the FRET signal (Figs. 1b and 1d). However, cells expressing DR4-CFP and DR4-YFP exhibit a positive FRET signal (Fig. 1c), demonstrating that flow cytometric FRET analysis can accurately recapitulate the known biochemical interactions of these receptors (Chan et al., 2000).
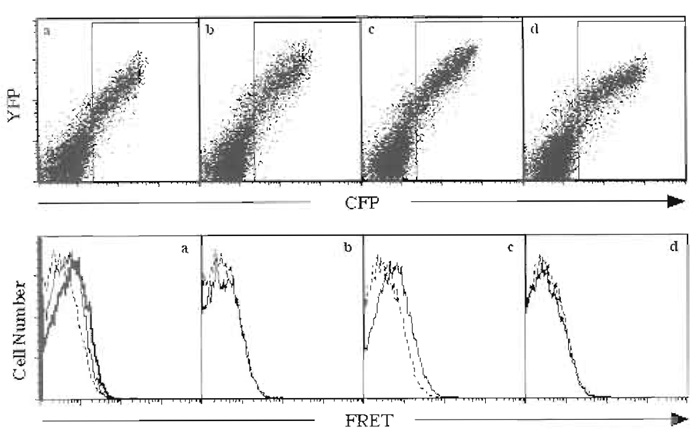 |
| FIGURE 1 HEK 293T cells were transfected using Fugene 6 with (a) p60-CFP and p60-YFP, (b) p60-CFP and DR4-YFP, (c) DR4-CFP and DR-YFP, or (d) DR4-CFP and p60-YFP. (Top) Live cells were analyzed for their expression of CFP and YFP. Cells that express both CFP and YFP were gated (rectangular box) and analyzed for FRET. (Bottom) Histogram overlays of control cells expressing only CFP (dotted lines) or cells expressing CFP and YFP (solid lines) show that only (a) p60-CFP and p60-YFP or (c) DR4-CFP and DR4-YFP exhibited FRET. (a) The heavy line shows the increased FRET when the ligand TNF was added to the cells. |
IV. COMMENTS
In addition to the FACS Vantage SE flow cytometer, other models of flow cytometers can be used for flow cytometric FRET analysis if the proper lasers are installed. For example, the LSRII flow cytometer from BD Bioscience can be adapted for FRET analysis if equipped with a violet laser for excitation at 405nm. Other methods of detecting molecular interactions using the concept of FRET include fluorescence lifetime imaging microscopy and microscopic photobleaching (Bastiaens and Squire, 1999; Miyawaki and Tsien, 2000).
IV. PITFALLS
- FRET is sensitive to the distance and orientation of the fluorescence donor and acceptor (Miyawaki and Tsien, 2000; Pollok and Heim, 1999; Truong and Ikura, 2001). Moreover, the spacer length between the CFP or YFP moieties and the protein of interest can also affect the efficiency of FRET (Chan et al., 2001). Therefore, it is sometimes necessary to test multiple fusion constructs to find the optimal fusions for FRET analysis.
- Low transfection efficiency is detrimental in FRET analysis. For 293T cells, Fugene 6 is the transfection reagent of choice because of its consistency in yielding high protein expression. Cell culture conditions can also be significant in the success of transfections. Typically, seeding 2.5 × 105 cells on 12-well plates will yield a 70-80% confluent culture after a 16- to 20-h incubation, which is ideal for high transfection efficiency.
- To optimize the FRET signal, it is necessary to have the fluorescence acceptor molecule in slight excess relative to the fluorescence donor molecule. Therefore, different ratios of CFP to YFP plasmids should be tested to determine the optimal ratio that will yield maximal FRET signals.
References
Bastiaens, P. I., and Squire, A. (1999). Fluorescence lifetime imaging microscopy: Spatial resolution of biochemical processes in the cell. Trends Cell Biol. 9, 48-52.
Chan, F. K., Siegel, R. M., Zacharias, D., Swofford, R., Holmes, K. L., Tsien, R. Y., and Lenardo, M. J. (2001). Fluorescence resonance energy transfer analysis of cell surface receptor interactions and signaling using spectral variants of the green fluorescent protein. Cytometry 44, 361-368.
Chan, F. K. M., Chun, H. J., Zheng, L., Siegel, R. M., Bui, K. L., and Lenardo, M. J. (2000). A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science.
Forster, T. (1946). Energiewanderung und fluoreszenz. Naturwissenschaften 6, 166-175.
Forster, T. (1948). Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. (Leipzig) 2, 55-75.
He, L., Olson, D. P., Wu, X., Karpova, T. S., McNally, J. G., and Lipsky, P. E. (2003). A flow cytometric method to detect protein-protein interaction in living cells by directly visualizing donor fluorophore quenching during CFP → YFP fluorescence resonance energy transfer (FRET). Cytometry 55A, 71-85.
Kanno, T., Kanno, Y., Siegel, R. M., Jang, M. K., Lenardo, M. J., and Ozato, K. (2004). Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. MoI. Cell 13, 33-43.
Miyawaki, A., and Tsien, R. Y. (2000). Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327, 472-500.
Pollok, B. A., and Heim, R. (1999). Using GFP in FRET-based applications. Trends Cell Biol. 9, 57-60.
Siegel, R. M., Frederiksen, J. K., Zacharias, D. A., Chan, F. K. M., Johnson, M., Lynch, D., Tsien, R. Y., and Lenardo, M. J. (2000). Fast preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science.
Stryer, L. (1978). Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 47, 819-846.
Truong, K., and Ikura, M. (2001). The use of FRET imaging microscopy to detect protein-protein interactions and protein conformational changes in vivo. Curr. Opin. Struct. Biol. 11, 573-578.
Tsien, R. Y. (1998). The green fluorescent protein. Annu. Rev. Biochem. 67, 509-544.
Yamagoe, S., Kanno, T., Kanno, Y., Sasaki, S., Siegel, R. M., Lenardo, M. J., Humphrey, G., Wang, Y., Nakatani, Y., Howard, B. H., and Ozato, K. (2003). Interaction of histone acetylases and deacetylases in vivo. Mol. Cell Biol. 23, 1025-1033.
Zahavy, E., Fisher, M., Bromberg, A., and Olshevsky, U. (2003). Detection of frequency resonance energy transfer pair on doublelabeled microsphere and Bacillus anthracis spores by flow cytometry. Appl. Environ. Microbiol. 69, 2330-2339.
Support our developers




