DNA Replication-Dependent Chromatin Assembly System
I. INTRODUCTIONChromatin is a periodic structure made up of repeating, regularly spaced subunits, the nucleosomes. Each nucleosome includes a core octamer of two molecules each of histone proteins H3, H4, H2A, and H2B, around which the DNA is wrapped almost twice. The assembly and structure of chromatin modulate the accessibility of proteins to the genome and hence regulate all processes that utilize the DNA. Analyses of transcription, DNA replication, and repair from in vitro-assembled chromatin templates reveal a wealth of information not provided by studies on naked DNA templates, as the chromatinized template more accurately reflects the state of the genome in the cell. This article outlines and compares systems for the assembly of recombinant DNA templates into arrays of nucleosomes in vitro to enable the reader to select the appropriate chromatin assembly system for their experimental question and resources. In addition, this article describes the DNA replication-coupled chromatin assembly system used in our laboratory and directs the reader to recent method chapters providing excellent descriptions of the other chromatin assembly systems. Chromatin assembly systems fall into two classes: "defined" systems where all the components are purified or recombinant and "cell-free" systems where the components required for assembly are supplied by a crude extract. The cell-free chromatin assembly systems can be further subcategorized into replication-independent systems and DNA synthesiscoupled systems. Defined chromatin assembly systems should be used if the experiment requires that any component of the chromatin assembly reaction or the resulting chromatin be manipulated, such as the nature of the histone proteins. Defined chromatin assembly systems fall into two classes: histone transfer systems and ATP-dependent chromatin assembly systems.
A. Defined Chromatin Assembly Systems for Histone Transfer
The core of the chromatin assembly reaction is the transfer of histone proteins to DNA, as mediated by a histone transfer vehicle or histone chaperone. If DNA and histones are combined in the absence of a histone transfer vehicle at physiological salt conditions, an undefined insoluble aggregate is obtained due to the intrinsic electrostatic attraction between the DNA and the highly basic histone proteins. A histone transfer vehicle binds to the histones and facilitates the ordered formation of nucleosomes from histones and DNA by reducing their inherent affinity for each other.
A variety of histone transfer vehicles can be used in vitro to assemble chromatin, including polyanions such as polyglutamate and counterions such as sodium chloride. The observation that high salt concentrations (>1.2M NaCl) dissociate nucleosomes led to the discovery that this process was reversible by the progressive dialysis of mixtures of DNA and histones from high to low salt (Fig. 1A) (Lee and Narlikar, 2002). Chromatin assembly by salt dialysis is the simplest and cleanest way to form nucleosomes and has been invaluable for structural analyses of single nucleosomes (mononucleosomes; Luger et al., 1999). However, arrays of nucleosomes made by salt dialysis are very close packed, with a nucleosome repeat length of around 150-160bp as compared to that of 180- 190bp for nucleosomes in vivo (nucleosome repeat length is the center-to-center distance between adjacent nucleosomes; Fig. 1). This unphysiological closepacked characteristic of the salt dialysed chromatin can be overcome by incubation with an ATP-dependent chromatin remodeling factor (see later; Fig. 1C). Salt dialysis can yield nucleosomal arrays with more physiological spacing when DNA templates that comprise tandem arrays of nucleosome positioning sequences derived from the sea urchin 5S RNA gene are used (Carruthers et al., 1999). However, the use of nucleosome positioning sequences places a sequence limitation on the DNA template and does not reflect the situation in vivo where nucleosomes are generally not positioned by the underlying DNA sequence.
Histone transfer in vitro can also be achieved by physiologically relevant histone chaperones, such as nucleosome assembly protein 1 (NAP-l), chromatin assembly factor 1 (CAF-1), and antisilencing function 1 (ASF1) or by histone storage proteins such as nucleoplasmin and N1/N2. Histone chaperonemediated chromatin assembly (in the absence of an ATP-dependent chromatin remodeling factor) generally results in the unphysiological random spacing of nucleosomes along the DNA (Fig. 1B). The major applications for nucleosomal arrays assembled by histone transfer are for protein interaction analyses and for testing the ability of chromatin remodeling factors to convert irregularly spaced or close-packed nucleosomes into physiologically spaced, regular nucleosomal arrays.
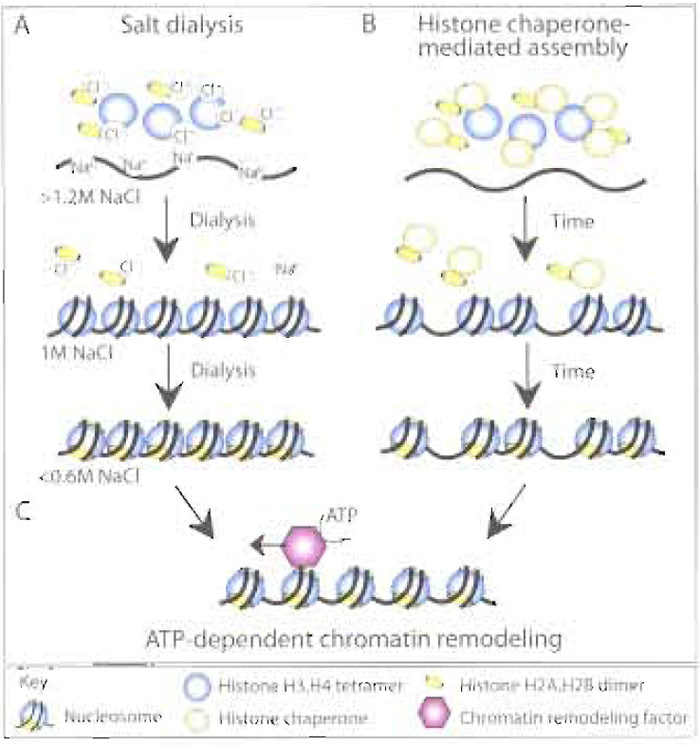 |
| FIGURE 1 Overview of chromatin assembly systems. Histones can be deposited onto DNA to form nucleosomes in vitro via (A) the sequential reduction in the salt (NaCl) concentration via dialysis, resulting in regular, but close packed arrays of nucleosomes, or (B) histone chaperone-mediated histone deposition onto DNA, resulting in irregular arrays of nucleosomes. (C) The presence of an ATP-dependent chromatin remodeling activity, either during or after histone deposition, will result in regular, physiological spacing of the nucleosomes. |
B. Defined ATP-Dependent Chromatin Assembly Systems
In order to generate regularly spaced arrays of nucleosomes that resemble chromatin in vivo, an ATPdependent chromatin remodeling factor is required in addition to a histone chaperone (Fig. 1C). Chromatin remodeling factors, e.g., ATP-utilizing chromatin assembly and remodeling factor (ACF) and chromatin accessibility complex (CHRAC), are multisubunit protein complexes that use the energy from ATP hydrolysis to space nucleosomes along the DNA template (Fig. 1C). These ATP-dependent chromatin assembly factors were identified by fractionation of Drosophila embryo chromatin assembly extracts and function together with histone chaperones, such as NAP-1 or CAF-1 (Fyodorov and Levenstein, 2002). A human ATP-dependent chromatin assembly factor, termed remodeling and spacing factor (RSF), appears to act as both the histone chaperone and the ATPdependent chromatin remodeling factor during chromatin assembly in vitro (Loyola et al., 2001). These defined ATP-dependent chromatin assembly systems provide powerful approaches to generate regular nucleosomal arrays with physiological nucleosomal spacing, albeit at the expense of time-consuming purification of the chromatin assembly factors and histones.
C. Generation of Reagents for Defined Chromatin Assembly Systems
In order to use defined chromatin assembly systems, it is necessary to use purified or recombinant chromatin assembly factors and histones. Recombinant histone chaperones and ATP-dependent chromatin remodeling factors are generated by affinity purification following baculovirus-mediated expression in Sf9 cells (Fyodorov and Levenstein, 2002). Core histones (H3, H4, H2A, and H2B) can be purified from a range of organisms using established protocols (Schnitzler, 2002). In addition, recombinant Xenopus, Drosophila, and yeast core histones can be expressed and purified from Escherichia coli, allowing assembly of chromatin lacking posttranslational modifications or with specific histone mutations (Lee and Narlikar, 2002; Levenstein and Kadonaga, 2002; Luger et al., 1999; White et al., 2001). For those with time constraints, individual recombinant Xenopus core histones can be purchased from Upstate Biologicals and individual purified core histones from calf thymus can be purchased from Roche Applied Science. Due to the highly conserved nature of chromatin assembly factors and nucleosome structure across eukaryotes, it is not generally necessary to adhere to species specificity within the chromatin assembly machinery, histones, and the subsequent application of the chromatin templates. For example, there is no gross functional or structural difference in the chromatin assembled from yeast, human, or Drosophila core histones using human, Drosophila, or yeast chromatin assembly factors.
D. Cell-Free Replication-Independent Chromatin Assembly Systems
Cell-free chromatin assembly systems were developed prior to the defined ATP-dependent chromatin assembly systems, but are still the system of choice for many researchers. Technically, chromatin assembly in cell-free systems requires less empirical optimization than defined ATP-dependent chromatin assembly systems, while still generating high-quality regular arrays of nucleosomes with physiological spacing in an ATP-dependent manner. Extracts remain a requirement for the analysis of processes coupled to chromatin assembly, such as the assembly of chromatin during ongoing DNA synthesis in vitro. Limitations of cell-free chromatin assembly systems include the inability to fully control the nature of histones and nonhistone proteins incorporated into the chromatin or the chromatin assembly factors used for assembly, as these are contained within the cell-free extract. Conversely, the crude nature of these extracts can be an advantage if, for example, unknown factors are required to facilitate subsequent transcriptional activation from the chromatin template.
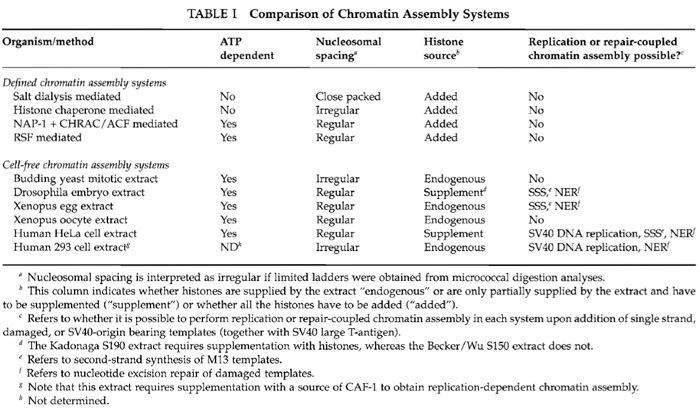
Cell-free extracts for generating physiologically spaced, regular arrays of nucleosomes have been derived from Xenopus laevis eggs and oocytes, Drosophila melanogaster embryos, and human tissue culture cells by high-speed (HS) centrifugation (Table I) (Bonte and Becker, 1999; Fyodorov and Levenstein, 2002; Gruss, 1999; Tremethick, 1999). The realization that the early developmental stages of Xenopus and Drosophila contain stockpiles of the components necessary to assemble the rapidly dividing DNA into chromatin was key to the development of highly active chromatin assembly extracts. For transcription analyses, the DNA template is usually assembled into chromatin using these extracts, followed by the addition of transcription factors or a transcription extract. Chromatin templates generated from the Xenopus egg HS extract are inherently flawed for subsequent transcriptional analyses due to the abundance of variant linker histone B4 and transcription factors in the extract. In fact, assembly of transcription complexes competes with chromatin assembly in the Xenopus egg HS extract. In general, the Xenopus egg HS extract is used for DNA synthesis-coupled chromatin assembly, whereas the Xenopus oocyte extract (S150; where S refers to the sedimentation coefficient) is used for assembling templates into chromatin for their subsequent transcriptional analyses (Gaillard et al., 1999; Tremethick, 1999). The Drosophila S150 and S190 embryo extracts (Bonte and Becker, 1999; Fyodorov and Levenstein, 2002) have been widely used to assemble chromatin templates for subsequent analyses of transcriptional regulation, recreating many of the features of transcriptional regulation in vivo (Robinson and Kadonaga, 1998). These extracts also contain factors that can modify subsequent transcription reactions, such as chromatin remodeling activities, and therefore it may be desirable to purify the chromatin templates by sedimentation centrifugation or size exclusion prior to transcription analysis. Removal of nonhistone proteins from the chromatin may be achieved by treatment with Sarkosyl prior to purification of the chromatin template (Mizuguchi and Wu, 1999). However, defined chromatin assembly systems are the ideal method for complete control of the subsequent transcription reaction on chromatin templates.
A chromatin assembly extract has been developed from Saccharomyces cerevisiae that enables the powerful combination of genetics and biochemistry (Table I). This yeast extract also supports transcriptional activation and can be isolated from any yeast strain that has been altered by genetic manipulations. This enables investigation of the effect of specific mutations on chromatin assembly and transcriptional regulation of the in vitro-assembled chromatin templates (Schultz, 1999). However, this extract is limited by its inability to generate extensive regular arrays of nucleosomes (Table I).
 |
E. DNA Synthesis-Coupled Chromatin Assembly Systems
In the cell, the majority of chromatin assembly is coupled to ongoing DNA replication and repair. The process of chromatin assembly coupled to doublestrand DNA replication has been recreated in vitro using extracts from human tissue culture cells (Table I). In addition, Drosophila, human cell, and Xenopus egg HS extracts can mediate nucleotide excision repaircoupled chromatin assembly on damaged templates and second-strand synthesis-coupled chromatin assembly on M13 templates (Bonte and Becker, 1999; Gaillard et al., 1999). The human 293 cell extract has been widely used to study the process of DNA replication coupled-chromatin assembly using an SV40 origin to initiate replication via the method described in this article (Fig. 2). In this system, DNA replication occurs on only a portion of the total DNA templates, allowing comparison of chromatin assembly on the newly replicated DNA (labelled with a radioactive nucleotide) to chromatin assembly on the total DNA (Fig. 2). As such, this system has led to the identification and characterization of histone chaperones that mediate chromatin assembly onto newly synthesized DNA, such as CAF-1 and ASF1 (Smith and Stillman, 1989; Tyler et al., 1999). Furthermore, the SV40 DNA template can be preassembled into chromatin using Drosophila and Xenopus chromatin assembly extracts and then replicated using human DNA replication extracts (Gruss, 1999). Refinement of this approach may recreate epigenetic inheritance of chromatin structures in vitro.
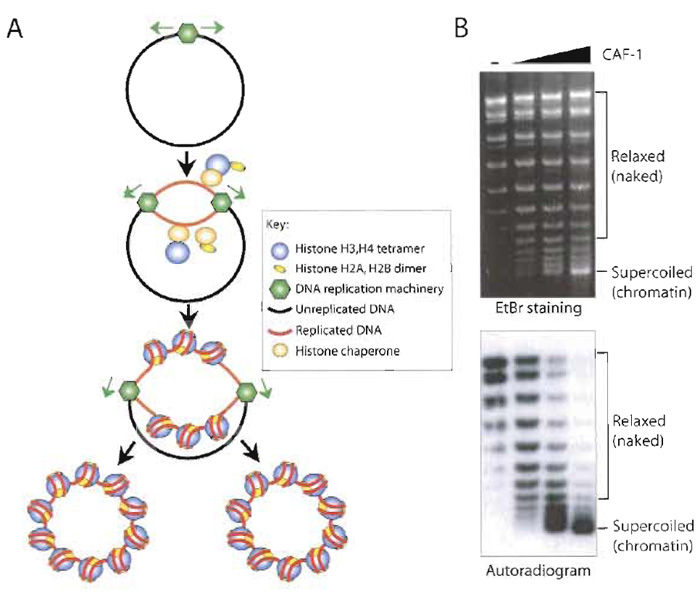 |
| FIGURE 2 DNA replication-coupled chromatin assembly. (A) Overview of the SV40 T-antigen-driven replication assay in a human cell-free extract coupled to chromatin assembly. The newly replicated DNA (red) becomes labelled by the incorporation of a radioactive nucleotide. (B) Example of supercoiling analysis of the products of DNA replication-coupled chromatin assembly. By comparison of EtBr staining and autoradiogram of the same agarose gel, preferential assembly (supercoiling) of the replicated DNA into chromatin is apparent upon addition of CAF-1. |
F. Evaluation of in Vitro.Assembled Chromatin
Once assembled, it is important to check the extent of nucleosome assembly and the regularity of nucleosomal spacing. The extent of chromatin assembly can be measured by DNA supercoiling analysis, as the formation of each nucleosome introduces one negative supercoil into covalently closed circular DNA. The number of negative supercoils therefore corresponds to the number of nucleosomes assembled and is measured after deproteinization by agarose gel electrophoresis. Plasmid supercoiling by an extract cannot always be attributed to chromatin assembly, as these extracts potentially contain unrelated DNA supercoiling activities. Therefore, it is necessary to confirm that the supercoiling is due to chromatin assembly, usually by the micrococcal nuclease digestion assay. In this assay, the chromatin is partially digested by micrococcal nuclease, which cleaves both strands of DNA between the nucleosomal cores. The resulting chromatin fragments are deproteinized and the DNA is resolved by agarose gel electrophoresis. A DNA ladder derived from the chromatin fragments is observed if the assembly was efficient and the nucleosomes were regularly spaced, and the size of the DNA fragments allows the nucleosome repeat length to be estimated.
II. MATERIALS AND INSTRUMENTATION
Plasmid template carrying SV40 origin of DNA replication [we use pSV011 (Stillman, 1986)]. SV40-T antigen, affinity purified (Simanis and Lane, 1985) or purchased (Cat. No. 5800-01) from CHIMERx. Spinner-adapted version of HEK293 cells (Cat. No. CRL1573) is from ATTC. Joklik's modification of MEM (Cat. No. 1032324) and sodium butyrate (Cat. No. 218453) are from ICN. Fetal bovine serum (Cat. No. 16000-044) is from Life Technologies Inc. Ribonucleotides (ATP, CTP, GTP, UTP, Cat. Nos. E6011, E6021, E6031, E6041) and deoxyribonucleotides (dATP, dCTP, dGTP, dTTP, Cat. No. U1330) are from Promega. Filters (0.22 µm) (Cat. No. GSWP04700) are from Millipore. A 40-ml Wheaton dounce homogenizer with B pestle (Cat. No. 06-435-C), Corex 8445 tubes (Cat. No. 05-566- 55), agarose (Cat. No. BP1356), chloroform (Cat. No. C298), EDTA (Cat. No. S311), dithiothreitol (DTT, Cat. No. BP172-25), glycerol (Cat. No. G3320), HEPES (Cat. No. BP310), magnesium chloride (MgCl2, Cat. No. M33), phenol: chloroform:isoamylalcohol 25:24:1 (Cat. No. BP17521), potassium chloride (KCl, Cat. No. P217), potassium hydroxide pellets (KOH, Cat. No. P250), potassium phospate monobasic (KH2PO4 Cat. No. P285), proteinase K (Cat. No. BP1700), sodium acetate trihydrate (NaOAc, Cat. No. S209), sodium chloride (NaCl, Cat. No. S271), sodium phosphate dibasic (Na2HPO4·7H2O Cat. No. S373), sodium phosphate monobasic (NaH2PO4 Cat. No. S369), Tris base (Cat. No. BP152), xylene cyanol FF (XC, Cat. No. BP565), and X-ray film (Cat. No. 05-728-24) are from Fisher. Boric acid (Cat. No. B0252), bromphenol blue (BPB, Cat. No. B-6131), creatine phosphate (Cat. No. P7936), creatine phosphokinase (CPK, Cat. No. C3755), ethidium bromide (EtBr, Cat. No. E-8751), glycogen (Cat. No. G0885), IGEPAL (NP40, Cat. No. I3021), RNase A (Cat. No. R6513), and sodium hydroxide pellets (NaOH, Cat. No. S-0899) are from Sigma. Sodium dodecyl sulphate (SDS, Cat. No. 161-0302) is from Bio-Rad. [α-32P]dATP 3000Ci/mmol, 10µCi/µl (Cat. No. BLU512H) is from Dupont NEN. Complete protease inhibitor cocktail (Cat. No. 1836153) is from Roche.
A 37°C tissue culture incubator (CO2 not required) and a magnetic stir plate for spinner flasks (Thermodyne Cat. No. 45700) are from Fisher. The 6-liter spinner flask (Cat. No. 1965-06000) is from Bellco. Refrigerated centrifuge Sorvall RC-5B with SS34, HB6, and GSA rotors (or equivalent). Beckman ultracentrifuge with Ti70 rotor. Rotary Speed-Vac, gel dryer, and -80°C freezer are from Thermo Savant (or similar). Agarose gel electrophoresis equipment, Gibco H5 system (or similar). Liquid nitrogen tank. Eppendorf Model 5415D microcentrifuge (or similar).
III. PROCEDURES
A. Preparation of DNA Replication- Dependent Chromatin Assembly Extract
These procedures are modified from those of Stillman (1986).
Solutions
- Phosphate-buffered saline (PBS): 136mM NaCl, 2.7 mM KCl, 3.4 mM Na2HPO4, and 1.47 mM KH2PO4. For 100ml of 1X PBS, combine 90 ml MilliQ or doubledistilled water (ddH2O) and 10ml 10X PBS, use within days. For 1 liter of 10X PBS, to 900ml ddH2O add 80g NaCl, 2g KCl, 9.2g Na2HPO4·7H2O, and 2g KH2PO4. Stir until dissolved and then add ddH2O to 1-liter final volume. Autoclave and store 10X PBS at 4°C.
- 1M HEPES-KOH, pH 8.0: For 1 liter, to 600ml ddH2O add 238.3 g HEPES. Once dissolved, adjust pH to 8.0 using KOH pellets and add ddH2O to 1-liter final volume. Pass through 0.22-µm filters. Store in a sterile bottle at 4°C.
- 3 M KCl: For 1 liter, to 700ml ddH2O add 223.68 g KCl. Once dissolved, add ddH2O to 1-liter final volume. Autoclave and store at room temperature.
- 1M MgCl2: For 1 liter, to 700ml ddH2O add 203.3g MgCl2. Once dissolved, add ddH2O to 1-liter volume. Autoclave and store at room temperature.
- 0.5M DTT: For 50ml, dissolve 3.85g DTT in 50ml ddH2O and store at -20°C.
- 1M sodium phosphate, pH 7.2: Dissolve 26.807g NaH2PO4 in 70ml ddH2O and adjust volume to 100ml once dissolved. Dissolve 13.8 g Na2HPO4·7H2O in 80ml ddH2O and adjust volume to 100ml once dissolved. Combine the two solutions together (approximately 68 ml :31 ml, respectively) until pH 7.2 is reached. Store at room temperature.
- 1M Na butyrate: 1M Na butyrate and 20mM sodium phosphate, pH 7.2. For 100ml, dissolve 11 g butyric acid (sodium salt) in 80ml ddH2O and then pH to 7.2 with NaOH. Next, add 2ml 1M sodium phosphate, pH 7.2. Add ddH2O to 100-ml final volume. Store at 4°C. This solution has an unpleasant odour.
- Hypotonic buffer: 20mM HEPES-KOH, pH 8.0, 5mM KCl, 1.5mM MgCl2, and 0.1mM DTT. For 100ml, add 2ml 1M HEPES-KOH stock, 0.16ml 3M KCl, 150µl 1M MgCl2, and 20µl 0.5M DTT (stored at -20°C. Bring to volume with ddH2O. Prepare fresh and chill on ice.
- 5M NaCl: For 1 liter, to 700 ml ddH2O add 292.2 g NaCl. Once dissolved, add ddH2O to 1-liter final volume. Autoclave and store at room temperature.
Steps
- Grow 4 liters of 293 cells in Joklik's modification of MEM plus 5% bovine calf serum in a 6-liter spinner flask to 6 × 105 cells/ml at 37°C. Note that because this medium is not buffered by sodium bicarbonate, you can use a sealed spinner in a 37°C room if necessary.
- Spin down 293 cells in six GSA bottles at 2500rpm at 4°C for 10min with brakes off. Pour off and discard the supernatant.
- Repeat step 2 using the same GSA bottles until all cells are pelleted. Keep the bottles on ice whenever possible. Perform all the following procedures in a 4°C room.
- Resuspend cells in the liquid remaining in the GSA bottles and transfer to an SS34 tube on ice. Wash the remaining cells out of the GSA bottles with chilled PBS and pool into the SS34 tube.
- Centrifuge at 2500rpm at 4°C for 3min and then slowly pour off and discard the supernatant.
- Add 20ml of chilled PBS to the cell pellet and resuspend by gentle pipetting with a plastic 25-ml pipette.
- Centrifuge cells again for 3 min and slowly pour off and discard the supernatant.
- Estimate the volume of packed cells in each tube in order to determine the volume of hypotonic buffer to be added later (by pipetting water into another SS34 tube and estimating the pellet volume by comparison, usually approximately 7- 8ml).
- To 100 ml hypotonic buffer, add 0.5 ml of 1M Na butyrate (to 5mM final) and protease inhibitor cocktail tablets.
- Add 20ml of hypotonic buffer (with additives) to cells and resuspend by gentle pipetting.
- Centrifuge at 2500 rpm at 4°C for 5 min and remove and discard the supernatant using a pipette.
- Resuspend cells in 1 ml of hypotonic buffer for each milliliter of packed cell volume estimated in step 8. Resuspend cells by gentle pipetting. Incubate cells on ice for 10min.
- Transfer cell suspension to a 40-ml dounce homogenizer. Rinse SS34 tube with a small amount of hypotonic buffer and pour the rinse into the dounce homogenizer. Remove 10 µl of cell suspension for step 14.
- Dounce cells on ice for 25-30 passages with a "B" pestle. Remove 10µl of cell suspension, and compare cells before and after douncing using an optical microscope to estimate extent of lysis. If there are significant numbers of intact cells after douncing, dounce for a few more passages.
- Pour lysed cells into two prechilled 30-ml Corex tubes. Incubate on ice for 30min.
- Centrifuge in an HB6 rotor at 10,000 rpm at 4°C for 10min. Pour the supernatant into a 15-ml conical tube to estimate volume. Add NaCl to 0.1M final concentration (a 1/50 dilution of 5M NaCl) into the supernatant and mix gently.
- Divide solution between two ultracentrifuge tubes (Beckman Ti70) and centrifuge in a prechilled Ti70 rotor at 31,000rpm at 4°C for 60min.
- Transfer the supernatant to a new tube (it is okay if some lipids transfer over). Divide into 0.5-ml aliquots, freeze in liquid N2, and store at -80°C.
B. DNA Replication-Dependent Chromatin Assembly and Supercoiling Assay
Solutions
- 1M creatine phosphate: Add 1 g of creatine phosphate to 80 µl 1M HEPES-KOH, pH 8.0, and 3.92 ml ddH2O. Store at -80°C.
- 5X RM: 200mM HEPES-KOH, pH 8.0, 15mM ATP, 1 mM CTP, 1 mM GTP, 1 mM UTP, 0.125 mM dATP, 0.5mM dCTP, 0.5mM dGTP, 0.5mM dTTP, 200 mM creatine phosphate, 40 mM MgCl2, and 2.5 mM DTT. To make 1.2ml 5X RM, combine, in order, 426.5 µl ddH2O, 240 µl 1M HEPES-KOH, pH 8.0, 180 µl 100mM ATP, 12µl 100mM CTP, 12µl 100mM GTP, 12 µl 100 mM UTP, 1.5 µl 100 mM dATP, 6 µl 100 mM dCTP, 6 µl 100 mM dGTP, 6 µl 100 mM of dTTP, 240 µl 1M creatine phosphate, 28 µl 1M Mg2Cl, and 6 µl 0.5M DTT. Freeze in liquid N2 and store at -80°C.
- 2M Tris-HCl, pH 7.5: to make 2 liters, add 484.4 g Tris base to 1.5 liters of ddH2O. Adjust pH to 7.5 with HCl. Pass through a 0.22-µm filter and store in in sterile bottle at room temperature.
- CPK: 5mg/ml CPK, 50% glycerol, 25mM Tris-HCl, pH 7.5, 1 mM EDTA, 25 mM NaCl, and 1 mM DTT. To make 10ml CPK, add 50mg creatine phosphokinase to 4.785 ml ddH2O, 5 ml glycerol, 125µl 2M Tris-HCl, pH 7.5, 20µl 0.5M EDTA, 50µl 5M NaCl, and 20µl 0.5M DTT. Make 10-µl aliquots, freeze in liquid N2, and store at -80°C.
- Master mix: For each of x reactions (x = n + 1, where n is the number of reactions being performed), combine on ice in the following order: 5µl 5X RM, 0.1 µl CPK, 1 µl template DNA (at 50ng/µl), 1 µl SV40 T-antigen, and 0.1 µl [α-32P]dATP. Use immediately.
- 0.5M EDTA, pH 8.0: For 1 liter, to 700ml ddH2O add 186.12g EDTA. Add NaOH pellets until white emulsion begins to clear. Adjust pH to 8.0 with NaOH pellets. Add ddH2O to 1-liter final volume. Autoclave and store at room temperature.
- 25 mM buffer A: 18.75 mM Tris-HCl, pH 7.5, 2.5% glycerol, 1 mM EDTA, and 0.125% NP-40. For 4ml, combine 3.45 ml ddH2O, 37.5 µl 2M Tris-HCl, pH 7.5, 500 µl glycerol, 10 µl 0.5M EDTA, and 4 µl NP-40. Store at 4°C.
- 10 mg/ml RNase A: Dissolve 1 g RNase A in 100 ml ddH2O. Make aliquots and store at -20°C.
- EDTA/RNase buffer: For 1 ml, combine 935µl ddH2O, 50µl 0.5M EDTA, pH 8.0, and 15µl 10mg/ml RNase A. Use immediately.
- Glycogen stop buffer: 20 mM EDTA, 1% (w/v) SDS, 0.2M NaCl, and 250µg/ml glycogen. For 50ml, combine 45.5 ml ddH2O, 2 ml 0.5M EDTA, 500 mg SDS, 2ml 5M NaCl, and 12.5mg glycogen. Store at room temperature.
- Proteinase K: 2.5mg/ml proteinase K, 10mM Tris-HCl, pH 7.5, and 1 mM EDTA. To make 10 ml, dissolve 25mg proteinase K in 9.9ml ddH2O, 50µl 2M Tris-HCl, pH 7.5, and 20 µl 0.5M EDTA. Make aliquots and store at -20°C.
- 3M NaOAc: For 1 liter, to 500ml ddH2O add 408.24g sodium acetate trihydrate. Once dissolved, add ddH2O to 1-liter final volume. Autoclave and store at room temperature.
- 1X TBE: 89mM Tris, 89mM boric acid, and 2.5 mM EDTA. For 1 liter of 1X TBE, combine 900ml ddH2O and 100ml of 10X TBE, For 1 liter 10X TBE, to 600ml ddH2O add 108 g Tris base, 55 g boric acid, and 7.49 g EDTA. Stir until dissolved and then add ddH2O to 1-liter final volume. Store at room temperature. Discard solution when white particles appear.
- 1X TBE gel-loading buffer: 89mM Tris, 89mM boric acid, 2.5 mM EDTA, 6% glycerol, 0.05% BPB, and 0.05% XC. For 50ml of 1X TBE gel loading buffer, combine 15 ml ddH2O, 15 ml glycerol, 25 ml 10X TBE, 0.125g BPB, and 0.125g XC. Store at 4°C.
- 10mg/ml EtBr: Add 0.5g EtBr (weighed in a fume hood) to 50ml ddH2O. Store in the dark at 4°C.
Steps
- Thaw T-antigen, CPK, 293 extract, and samples being tested in a room temperature water bath and place on ice once thawed. Assemble master mix on ice. Set up replication reactions on ice by adding in order 7.8 µl 25 mM buffer A, 7.2 µl master mix, and 10 µl 293 replication extract. Mix by pipetting up and down. If samples are being tested for their effect on DNA replication and/or chromatin assembly, add the samples first and adjust the volume of 25 mM buffer A so that the reaction has a final volume of 25 µl. Discard remainder of CPK aliquot and master mix. Freeze all unused reaction components in liquid N2 and store at -80°C.
- Incubate replication reactions at 30°C for 90min.
- Stop reactions by the addition of 6.25µl EDTA/RNase buffer. Incubate at room temperature for 15 min.
- Add 95µl glycogen stop buffer and 5µl proteinase K. Incubate at 37°C for 15 min.
- Extract samples with 1 volume of phenol:chloroform: isoamyl alcohol (25:24:1) and then with 1 volume of chloroform:isoamyl alcohol (24:1).
- Precipitate DNA by addition of 1/10 volume of 3M NaOAc and 3 volumes of 100% EtOH. Invert to mix and microcentrifuge at full speed for 15 min.
- Remove and discard supernatant and wash pellets with 75% EtOH. Dry pellets in a Speed-Vac.
- Resuspend pellets in 8µl of 1X TBE gel loading buffer. Load into wells of 1% agarose gel in 1X TBE and run until BPB runs off the gel (35 V for 14 h).
- Stain gel in 500ml ddH2O + 30µl 10mg/ml EtBr for 30min. Destain in ddH2O for 30-60min and photograph.
- Dry gel on two layers of Whatman 3 MM paper overlaid with Saran wrap on a gel drier for 2h with heater set to 65°C. Expose gel to film.
Figure 2B shows the products of the DNA replication- coupled chromatin assembly reactions, where the top shows total DNA and the bottom shows the newly replicated DNA.
IV. COMMENTS
- For each batch of T-antigen, the optimal amount needs to be determined empirically by determining the lowest amount that gives the maximum amount of replication. Occasionally, the volume of each extract for chromatin assembly may need to be determined in a similar manner.
- The amount of sample to use in a typical reaction needs to be determined empirically, as addition of too much chromatin assembly factor (i.e., CAF-1, ASF1) will inhibit DNA replication. Because too much salt in the samples can interfere with replication efficiency, in general it is advisable to dialyze samples into 25 mM buffer A before replication analysis.
- Controls for replication dependence, as opposed to nucleotide excision repair of the nicked plasmid, include omission of T-antigen or use of a plasmid lacking the SV40 origin of DNA replication.
V. PITFALLS
In our hands, there is significant variation between different 293 extracts in terms of the amount of active CAF-1 and ASF1 that they contain, for unknown reasons. For example, we have generated extracts that do not require the addition of CAF-1 for chromatin assembly, only require the addition of CAF-1, or require the addition of both CAF-1 and ASF1, and we have even generated extracts that fail to replicate DNA! Therefore, each extract needs to be assessed by performing the replication reaction and supercoiling analysis with and without complementation by CAF- 1, ASF1, or crude extracts. This extract variability is not an issue if a crude extract (such as the cell-free chromatin assembly extracts described in Section I,C) are being used to supply chromatin assembly factors. In fact, the variability in the extracts has allowed the original biochemical identification of CAF-1 and ASF1 by complementation of the 293 extracts (Smith and Stillman, 1989; Tyler et al., 1999).
References
Bonte, E., and Becker, P. (1999). Preparation of chromatin assembly extracts from preblastoderm drosophila embryos. In "Methods in Molecular Biology" (P. B. Becker, ed.), pp. 187-194. Humana Press, Totowa.
Carruthers, L. M., Tse, C., Walter, K. P., and Hansen, J. C. (1999). Assembly of defined nucleosomal and chromatin arrays from pure components. Methods Enzymol. 19-34.
Fyodorov, D., and Levenstein, M. E. (2002). Chromatin assembly using Drosophila systems. In "Current Protocols in Molecular Biology" (E Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, K. Struhl, eds.), Unit 21.7. Wiley, New York.
Gaillard, P. H., Roche, D., and Almouzni, G. (1999). Nucleotide excision repair coupled to chromatin assembly. In "Methods in Molecular Biology" (P. B. Becker, ed.), pp. 231-243. Humana Press, Totowa.
Gruss, C. (1999). In vitro replication of chromatin templates. In "Methods in Molecular Biology" (P. B. Becker, ed.), pp. 291-302. Humana Press, Totowa.
Lee, K., and Narlikar, G. J. (2002). Assembly of nucleosomal templates by salt dialysis. In "Current Protocols in Molecular Biology" (E Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, K. Struhl, eds.), Unit 21.6. Wiley, New York.
Levenstein, M. E., and Kadonaga, J. T. (2002). Biochemical analysis of chromatin containing recombinant Drosophila core histones. J. Biol. Chem. 277, 8749-8754.
Loyola, A., LeRoy, G., Wang, Y. H., and Reinberg, D. (2001). Reconstitution of recombinant chromatin establishes a requirement for histone-tail modifications during chromatin assembly and transcription. Genes Dev. 15, 2837-2851.
Luger, K., Rechsteiner, T. J., and Richmond, T. J. (1999). Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 119, 1-16.
Mizuguchi, G., and Wu, C. (1999). Nucleosome remodeling factor NURF and in vitro transcription of chromatin. In "Methods in Molecular Biology" (P. Becker, ed.), pp. 333-342. Humana Press, Totowa.
Robinson, K. M., and Kadonaga, J. T. (1998). The use of chromatin templates to recreate transcriptional regulatory phenomena in vitro. Biochim. Biophys. Acta. 1378, M1-6.
Schnitzler, G. (2002). Isolation of histones and nucleosome cores from mammalian cells. In "Current Protocols in Molecular Biology" (E Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, K. Struhl, eds.), Unit 21.5. Wiley, New York.
Schultz, M. C. (1999). Chromatin assembly in yeast cell-free extracts. Methods 17, 161-172.
Simanis, V., and Lane, D. P. (1985). An immunoaffinity purification procedure for SV40 large T antigen. Virology 144, 88-100.
Smith, S., and Stillman, B. (1989). Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 58, 15-25.
Stillman, B. (1986). Chromatin assembly during SV40 DNA replication in vitro. Cell 45, 555-565.
Tremethick, D. J. (1999). Preparation of chromatin assembly extracts from Xenopus oocytes. In "Methods in Molecular Biology" (P. B. Becker, ed.), pp. 175-186. Humana Press, Totowa.
Tyler, J. K., Adams, C. R., Chen, S. R., Kobayashi, R., Kamakaka, R. T., and Kadonaga, J. T. (1999). The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402, 555-560.
White, C. L., Suto, R. K., and Luger, K. (2001). Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 20, 5207-5218.
Support our developers




