High-Speed Cell Sorting
I. INTRODUCTIONFlow cytometric sorting is an extremely versatile technology that has established itself as a cornerstone in biological research for the foreseeable future. The strength and long-term success of this approach to cell purification can be attributed to its highly quantitative, rapid, and serial nature of analysis. Each passing particle is individually interrogated for the presence or absence of potentially limitless combinations of light scattering and fluorescence parameters. If user-defined threshold values are met, desired cells are then isolated from a heterogeneous sample. As such, the cell sorter becomes a launching point for many downstream cellular and molecular investigations, as well as a powerful tool for the analysis of complex mixtures.
The modern age of discovery-based, system-wide approaches to research is contingent upon high throughput analysis, and cell sorters have had to evolve accordingly to keep pace with these changes. Aptly, the most significant improvements in sorting technology were catalyzed by large-scale DNA sequencing efforts such as the Human Genome Project (Van Dilla and Deaven, 1990). It was obvious that by providing chromosome-specific DNA libraries, cell sorters could accelerate as well as improve sequencing results. The problem, however, was that literally days of sorting would be necessary to isolate sufficient genetic material for sequencing [modern polymerase chain reaction (PCR) techniques had not yet been reported]. Ensuing advances in the fluidics, illumination, optics, computers, and electronics of sorters led to the development of several experimental instruments that could sort at rates nearly two orders of magnitude above existing machines, and eventually laid the groundwork for modern, highend, high-speed sorters (Peters et al., 1985; Gray et al., 1987).
It is important to stress that the term "high speed" does not refer to simply speeding up the sort process, but is the product of careful engineering of each individual component in order to optimize overall performance. Coupled with these developments, the newest instruments have also become modular, with openly accessible and changeable parts, resulting in greater flexibility and sophistication with simpler and more stable operation.
The continuing evolution of high-speed cell sorters illustrates how advances in scientific knowledge and laboratory techniques allow for novel applications of existing methodologies, whereas improvements in instrument design facilitate previously unapproachable experiments. Accordingly, we have divided this article into two main parts. The first presents ideas that will yield better results in any experimental setting, with focus geared toward cytometer setup and general instrumentation. The following sections discuss biological aspects of study design, particularly pertaining to newer applications that rely on the added features and throughput of high-speed machines. The latter protocols do not include the traditional bulk separation of biological material, but instead take advantage of the capacity to screen rapidly through extremely complex cell and molecular populations for the isolation of highly defined, rare events. Specifically, fluorescent reporter proteins have provided the cytometrist with a real-time window into gene expression and have become the focus of many of these experimental approaches. It is these newest applications of high-speed cell sorting coupled with our increasing ability to study fewer, more characterized cells that will have the greatest impact on basic science research and our knowledge of human biology.
II. HIGH-SPEED CELL SORTERS: TECHNIQUES FOR RELIABLE SORTING
High-speed cell sorters are currently available from a handful of manufacturers with varying specifications, capabilities, facility requirements, and user interfaces. The purchase of any one particular instrument will depend on the desires of individual laboratories and should come after careful consideration of these differences. Although there are a number of mechanisms for sorting cells into discrete populations, the most widely used method for high-speed cell sorting involves using a jet in air, where cells contained in charged droplets are deflected using a static electric field. While the discussion that follows is most relevant to these sorters, the majority of protocols will have broader application. This section outlines some suggestions for consistent, reliable cell sorting that can be instituted at any sorting laboratory.
A. Cell Sorter Maintenance
Successful high-speed cell sorting requires both careful preparation of the sample to be studied and maintenance of the instrument being used. In most cases, the upkeep of electronics, lasers, deflection plates, software, and other hardware is left to service professionals, while alignment of optics and maintenance of fluidics and associated systems remain the purview of the user. In practice, however, basic knowledge of all aspects of the sort process is needed to ensure optimal and consistent operation.
In cell sorters, the fluidics layout is composed of two subsystems: the sample injection system and the sheath fluid system that surrounds the sample core. Together, these two subsystems contain several feet of tubing, and it is into the interstices and cracks of the tubing and valves that contaminants collect and can affect sort purity. As a result, the tubing material should be chosen to be as inert as possible and changed on a regular basis. Likewise, valves and corners should be kept to a minimum. Typically, the sheath tubing, sample tubing, and associated in-line filters should be changed at least monthly. New tubing should be washed and flushed for at least 10min prior to use with a 2% bleach solution, followed by sterile sheath fluid. For cell sorting where even slight contamination can skew results greatly, such as cases in which the sorted fraction will be used for quantitative PCR, it is not farfetched for the sample tubing to be replaced at the start of each sort. For its combination of inertness and cost, materials such as FEP (Teflon) are a good choice for the sheath tubing. FEP is chemically inert to a wide range of materials, holds up well to both acids and alkalis, and is autoclavable. An alternative to FEP that has better mechanical properties, particularly with respect to plastic deformation, is Tygon S-50-HL surgical grade tubing. Like FEP, this material can be,sterilized by autoclaving (30min, 15psi, 250 F). Unfortunately, sterilization does not always free tubing of inert inorganic contaminants, and thorough flushing prior to each use is recommended. For sample injection tubing, an ideal choice is PEEK tubing. Like FEP, it is chemically inert and has good mechanical properties necessary for rigidity of the smaller diameter tubing.
At the start of each day, the sorter should be flushed for 10min with a 2% bleach solution through both the sample and sheath lines, again followed by sterile sheath fluid to remove the bleach. In a similar manner, the sample injection should be flushed between samples when cross contamination is an issue. Finally, at the end of each day of sorting, it is recommended to flush the sheath and sample lines with a 2% bleach solution for 10min followed by deionized water to reduce salt crystal accumulation and then air-dried. Attaching an empty sample tube and sheath container to the sorter and pressurizing each for a brief period can accomplish air drying of the sample and sheath lines. The tubing and the sheath containers should be stored dry in a clean place.
B. Sheath Fluid
It is often overlooked that cell samples can be sensitive to even minor changes in the composition of the sheath fluid. One may erroneously assume that the sample does not interact significantly with the sheath, as there is no mixing between the two (the flow at the sample injection is laminar) and ions and solutes can move only by diffusion. This is not always the case, and the sample often interacts significantly with the sheath fluid, as shown in Fig. 1. In Fig. 1, zymosan particles have been labeled with FITC, a fluorophore whose absorption cross section is pH sensitive at 488nm, but not at 458nm. In Fig. l a, fluorescence efficiency at 488nm (normalized by the fluorescence at 458nm) changes when the pH of the sample is altered, causing an apparent change in measured fluorescence and potentially altering experimental results. However, these changes are observed only when the pH of the sheath is altered in the same manner as the sample fluid and are no longer apparent without changes in sheath fluid pH (Fig. lb). Commercial sheath fluid can be obtained with various chemical properties and constituents that can potentially affect the characteristics of the sample being studied. It is necessary to be familiar with the exact makeup of any sheath selection and to determine its likelihood of either chemical or biological interaction with the experiment. A simpler and less expensive alternative is to prepare a 0.9% NaCl solution and filter sterilize it or to filter the solution and then sterilize by autoclaving. Filtration of self-prepared sheath will reduce the number of nozzle clogs dramatically.
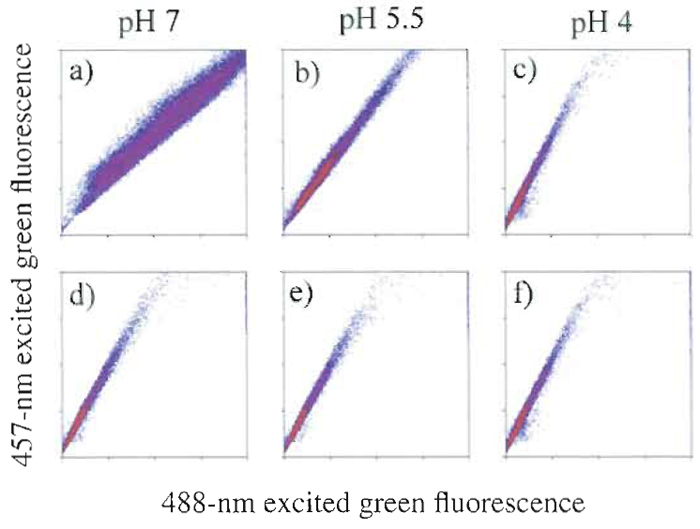 |
| FIGURE 1 FITC-labeled zymosan particles excited at two wavelengths. When excited with 488-nm light, FITC shows a pH-dependent fluorescence emission that is not seen when 457-nm light is used. Columns show the pH of the sample. In the top row, the pH of the sheath was adjusted to match that of the sample, but in the bottom row, it was maintained at pH 4. One can see that the pH of the sheath is a determinant for fluorescence efficiency. |
C. Sample Preparation and Determination of Sort Rate
High-speed cell sorters separate complex cell mixtures into constituent fractions so they can be studied in isolation. In the sorter, a jet of sheath fluid emanating from a nozzle surrounds the cell suspension coaxially. In order to separate events from one another, an acoustic vibration is coupled to the tip of the nozzle, and the vibrating nozzle leaves a trail of cyclical imprints onto the surface of the liquid. Because of surface tension energy, the cylinder of fluid is inherently less stable than a sphere of fluid, and soon the jet separates into regularly spaced droplets. Therefore, the cells, first carried by a cylinder of liquid, soon find themselves distributed randomly among the droplets. To separate the desired fraction of cells, an electrical charge may be applied to the droplets containing cells of interest as they separate from the main jet. In this manner, droplets with different cell types are directed toward collection vials by a static electrical field. On average, there are several empty droplets between each passing cell. As a result, the event rate (number of cells analyzed per second) should not be greater than the droplet formation rate. A typical droplet formation rate for a high-speed machine will be on the order of 50,000 drops/s, and the maximal throughput will be obtained if, on average, there is one cell per droplet. For this situation, however, statistics regarding the distribution of the cells among the droplets dictate that only 37% of the droplets will have one cell. The remainder of the droplets will be empty or contain more than one cell. Thus, the maximal sort rate becomes roughly 37% of the droplet formation rate; higher rates will cause too many cells to be lost due to coincident drop occupation (and hence, equivocal signal measurement), and lower rates will result in unnecessarily slow sorts. Without this information, standard coincidence rejection protocols require a 1.5 droplet interval between sortable events. As a result, we recommend limiting sort rates to roughly one-third of the droplet formation rate. Again, each sorting facility will have to perform a series of basic experiments to determine what is most appropriate for a given application. A more thorough derivation of the physics of drop formation and determinants of sort rate can be found in van den Engh (2000).
As a consequence of the desire to sort cells with maximal throughput, one must also determine the density of cells in the sample suspension. This consideration arises from needing to control the droplet occupancy rate (as described earlier) independent of the droplet formation rate and the sheath fluid pressure. Control of the sample injection rate is achieved by independently pressurizing the sample and the sheath fluid and altering the differential pressure between the two fluidic systems. There is a limit, however, to the degree one can increase the sample injection rate by increasing the sample pressure. Jet velocity is proportional to the square root of jet pressure, so large increases in pressure will result in relatively smaller changes in jet velocity. Furthermore, as the sample pressure continues to increase, a critical point will be reached that will disrupt the laminar injection of the sample into the core of sheath fluid. In this situation, the cells will not be hydrodynamically focused by the sheath fluid and will disperse throughout the fluid column. Cells would then encounter different laser light intensity as a function of their position in the sheath fluid, compromising measurements of fluorescence and scattered light from particles and resulting in decreased sort purity. To preserve fluidics integrity, the sample injection pressure should be within 1-3 psi of the sheath pressure. Additionally, it is difficult to control the event rate of a sample that has far too many cells per unit volume, as small changes in the differential pressure will result in uncontrollably large changes in the event rate. Although the desired density of the sample depends critically on the diameter of the sample injection tube (most are of the order of 125 µm), the majority of situations dictate sample densities of roughly 1-10× 106ml-1 with higher densities appropriate for smaller diameter sample injection tubes. Other factors to consider will depend on the inherent biological characteristics of the sample itself. For instance, certain cell lines are adhesive and may clump easier, making control of the sort rate difficult.
III. TROUBLESHOOTING
Cell sorters are complex machines and, as such, seemingly minor alterations or inconsistencies in set up and/or calibration can have an additive effect on sort purity. This section lists some common problems and suggested solutions.
A. Droplet Formation
Poor droplet formation, as evidenced by movement of the break-off point (the point at which the stream becomes discrete drops) with respect to the laser, will result in both poor yield and contamination. If the break-off point is not well stabilized, check to ensure that the nozzle is free of trapped air or debris and that the sheath fluid has equilibrated to the temperature of the room in which sorting is conducted. Other causes of poor droplet formation can be the presence of detergent in the system (which changes the surface tension energy of the fluid column) or a piezoelectric element that is poorly coupled to the nozzle.
B. High Background Event Rates
High background count rates (triggering of sort electronics without sample injection and/or event rates disproportionately above expected rates for a given sample) are generally the result of poorly maintained tubing or improperly prepared sheath fluid. These considerations are of particular importance when sorting or analyzing samples that are relatively dilute. We have found that a well-maintained sorter should have a background count of less than three particles per second. A good way to measure this is by using 1-µm beads and adjusting the forward and perpendicular scatter detector gains to one-fourth of their full range. After the sample injection valve is closed, one should then be able to differentiate background count rates from particles that are larger than 0.5µm. Should the background be unacceptably high, filtering of sheath fluid through a 0.22-µm filter or changing the sample and/or sheath fluid lines may be necessary.
C. Nozzle Clogs
Frequent nozzle clogs are usually the result of poor cleanup from a prior sort, suboptimal sample preparation, or inappropriate injection rate. Most commonly, sheath fluid left throughout the fluidics system from a previous sort dries and salt crystals are formed that clog the nozzle at the start of the following sort. Following the suggestions for flushing at the conclusion of each sort will remedy this issue. Additionally, one should examine the nozzle tip with a microscope to ensure that it is free of contamination and is uniformly smooth and patent. Certain samples will be more prone to causing clogs and, as such, the maximum injection pressure that will avoid clogs will have to be determined for each case.
The preceding points of discussion will vary to some degree among individual machines, laboratory settings, and instrument operators. In practice, machine start up, shut down, maintenance, sample manipulation, and so on, will all have an impact on overall, effective sort rate and general practice habits, being somewhat unique to each application. With consistent and thorough protocols for maintaining the sorting machinery, familiarization with the components of the sort process, and a systematic, skilled approach to troubleshooting, one can focus on the more exciting and challenging experimental aspects of high-speed cell sorting.
IV. STRATEGIES FOR EXPERIMENTAL DESIGN
While the original applications of high-speed cell sorting focused on the bulk purification of cellular and molecular material, modern biology has become increasingly focused on the minor nuances that distinguish one cell type from another. Although many biological experiments can still be approached through the analysis of cell populations, there is a growing need and improving ability to attain information from highly specified phenotypes, or even single cells. High-speed cell sorters are ideally suited for these applications because, by definition, a more highly classified cell will be present in less numbers, and it is often necessary to serially screen millions of individual events in a relatively short time period to find those particles of interest. Recent examples in the literature include the analysis of B cells from multiple myeloma patients to isolate subpopulations of cells with specific mutations in pathogenic genes (Kalakonda et al., 2001) or the haplotyping of individual sperm cells to look for trinucleotide repeat expansion in the Huntington's disease locus (Chong et al., 1997).
Beyond the study of naturally occurring variations and mutations, cell sorters have emerged as tools to screen engineered cell-based or solid support molecular libraries to select genetic clones and/or molecules of interest. Ingenious fluorescent-labeling schemes are employed to screen immensely complex libraries of particles to detect rare yet significant molecular processes such as enzyme activity or antibody binding and to monitor gene expression (Boder et al., 2000; Olsen et al., 2000). A revolution in molecular biology that is ideally suited to flow cytometry is the cloning of fluorescent reporter genes such as green fluorescent protein (GFP), discosoma Red (dsRed), and their variants (Chalfie et al., 1994; Matz et al., 1999). When fused to coding sequences of interest, these proteins generate fluorescent molecules from cells that express the cloned sequences in a dose-dependent fashion. Simple excitation of fluorescent proteins with a laser of appropriate wavelength results in the emission of a highly quantitative signal easily detectable by a flow cytometen Combinations of these proteins allow for the design of gene expression studies under a variety of experimental conditions. Cells expressing the desired proteins (and hence cloned sequences) can be separated in a cell sorter and interrogated in further studies.
V. PROCEDURES
A. Library Construction and Clone Selection
The ability to isolate useful DNA fragments from larger complex mixtures is critical for all of molecular biology. Because single molecules of DNA cannot be readily seen or handled, target DNA is ligated into a larger vector that can be manipulated and propagated in a host bacterial strain, most commonly E. scherichia coli. This construct usually consists of a plasmid or phage, but can be larger, such as with bacterial artificial chromosomes (BACs), or the entire bacterial chromosome. Well-described molecular biology techniques can be applied in this fashion to create highly diverse libraries rapidly with complexities greater than 109 and lie outside the scope of this article. With traditional methods, E. coli cells are grown on agar plates under antibiotic selection, with each colony representing a construct that has entered a bacterial cell successfully. Colonies with the desired genotype are then selected based on a colormetric phenotype and transferred to liquid media or streaked out on another agar plate for growth. Selected species can then be isolated efficiently with commercially available plasmid prep kits (Qiagen, Inc.) or by other established DNA extraction and amplification methods.
B. Cell Sorters as a Tool for Clone Selection and Genetic Screening
While the aforementioned approach is accurate and has been successfully employed for decades, cell sorters are ideally equipped to perform the same tasks in less time with added flexibility. At the most basic level, markers can be devised to simply indicate the presence or absence of a cloned insert. With more sophisticated study design and genetic manipulation, sorters become tools to potentially screen genomes of entire organisms in rapid fashion, providing a window into genetic regulation. Libraries can be screened and clones displaying a desired phenotype can be isolated, their DNA amplified, and eventually sequenced. As an example, an experiment could be fashioned to screen the entire Saccharomyces cerevisiae genome (~14Mbp) by generating 100-bp fragments placed strategically in relation to a reporter construct. High-end sorters would be capable of scanning the genome to 10X coverage for specific expression events in a matter of minutes. A similar approach was reported in a study by Barker et al. (1998) that generated a library of 200- to 1000-bp fragments of the Mycobacterium marinum genome fused to a promoterless copy of GFP. Like its counterpart Mycobacterium tuberculosis, this bacterium is able to avert the normal function of the immune system and survive within macrophages. By isolating phagosomes emitting green fluorescence and sequencing the regions of DNA upstream of GFP, they were able to determine which regions of the genome may be responsible for circumventing normal bacterial killing.
C. Fluorescence Encoding Strategies
The major challenge in experimental design is then to devise a strategy for expressing some characteristic of the cloned and/or expressed DNA as fluorescence. Because fluorescent proteins such as GFP and its variants can be coupled to genetic sequences and produced endogenously within bacteria without the need for added substrates, they become the obvious starting point. Basic approaches to clone selection indicate the presence of foreign DNA with the loss of a fluorescent signal. Inouye et al. (1997) developed a plasmid vector that directs the production of GFP under a constitutive promoter. When an insert is cloned into the vector, stop codons within the insert prevent the successful translation of GFP. Therefore, nonfluorescent colonies can be picked to obtain individual clones. In our experience, negative selection (based on the absence of fluorescence) can be a poor criterion for the analysis of individual cells in a cell sorter. Background particles are usually weakly fluorescent and, as such, it becomes difficult to segregate them from nonfluorescent (i.e., insert containing) bacteria. Additionally, a large degree of cell-to-cell variability exists that is widely determined by different sizes of bacterial cells and varying levels of protein expression. The combination of these factors makes it difficult to be certain that a nonfluorescent cell is not simply expressing a fluorescent marker at low levels and will result in sort contamination.
More complex schemes can be derived to represent unique characteristics of individual clones as positive fluorescence signals that result in sorts of higher purity. For instance, Olsen et al. (2000) created a scheme in which protease variants from a mutant library were expressed on the surface of E. coli. Functional enzyme mutants successfully cut a synthetic substrate bound to the E. coli membrane containing BODIPY and tetramethylrhodamine fluorophores connected by a linker with a specific target site. The tetramethylrhodamine acts as a quencher for the BODIPY dye by fluorescence resonance energy transfer. If the OmpT mutant on a particular cell cleaved the substrate, then the tetramethylrhodamine diffused away and BODIPY was no longer quenched. Sorting the fluorescent particles resulted in a 5000-fold enrichment for clones containing active variants of the protease.
As an additional example of immense potential for such techniques, Koo et al. (2004) devised a means to encode the activity of mRNA processing in E. coli as a fluorescent signal. A GFP construct was created as a fusion to a mRNA processing region. If functional mRNA processing was present, many of the GFP transcripts were degraded, leading to a low level of GFP fluorescence. If the ability to perform mRNA processing was disrupted, cells had high levels of GFP and were isolated. Using this method, over 60% of bacteria were highly fluorescent after four rounds of sorting.
VI. PRACTICAL CONSIDERATIONS
Once an algorithm for encoding biological function as a fluorescent signal has been conceived, it is vital to test the system under a variety of culture and cellular conditions. Often times, fluorescence from GFP and other fluorescent proteins is not visible in actively growing E. coli cultures. Because the rate of replication of E. coli is so high, any fluorescent protein that is produced is diluted continuously and, as such, fluorescence will not usually begin to appear until late log phase or stationary phase. To obtain higher cell-to-cell consistency of fluorescence, it is often helpful to grow the cells under conditions that repress fluorescent protein production during active growth. This can be achieved by cloning the fluorescent protein downstream from a tightly regulated promoter. If a lac promoter is used, growth in 0.5% glucose results in almost complete repression. In late log or stationary phase, cells can be resuspended in an equal volume of culture medium that does not repress protein production. With this approach, all cells begin producing fluorescent protein at approximately the same time and the rate of division has slowed, resulting in more consistent levels of fluorescent protein expression.
During testing, it is often convenient to use a fluorescence microscope with appropriate excitation and emission filters, as well as high-power objectives to image individual bacterial cells. Direct observation of cultured cells can be a quick method for assessing the levels of various fluorescent markers, as well as looking for undue elongation of cells that can indicate unhealthy cellular states. Only after thorough testing should bacterial cultures be inspected by flow cytometry. Typically, forward scatter is not an informative measurement to record but is an excellent parameter to trigger on as it can easily detect bacterial cells in a flow stream regardless of their orientation. This is a better method than triggering on fluorescence to prevent nonfluorescent cells from altering sort outcome.
A. Optical Setup and Instrument Settings
Fluorescence measurements can be performed with a variety of optical configurations. If a 488-nm laser is used to detect forward scatter, it is convenient to use this same laser for GFP measurements. Additionally, some variants of YFP (yellow mutant of GFP) and DsRed can be excited readily with a 488-nm beam. Other laser lines such as multiline UV or 514.5 nm can be used as needed. If multiple fluorescent proteins or markers are being utilized it is often efficient to use a dichroic beam splitter to efficiently separate fluorescence into short and long wavelength pathways. Further filtering with long-pass, band-pass, or shortpass filters can fine tune wavelength ranges to increase the number of measured parameters and to minimize cross talk between signals.
Bacterial cultures should be diluted approximately 200:1 in saline before injection into the flow cytometer. This can be variable depending on densities of cultures and cytometer configuration. The sample injection pressure on the flow cytometer should be adjusted for optimal event rate on each individual sorter as discussed previously. Test sorts should be performed using control cells first until the researcher attains some level of confidence that desired cell populations can be isolated successfully. In addition, these initial tests can often provide some estimate as to the enrichment that can be achieved with a particular fluorescence-encoding scheme. Tests should also be performed to assess the viability of bacteria after culturing and sorting. This can be achieved easily by sorting a particular number of cells into LB medium or saline and then serial dilutions of these sorted cells can be plated. After overnight growth, colony counts will provide a measure of viability. Optimally, 50-70% of injected cells should be viable. However, viability can drop below 1% depending on factors such as cell strain and culture conditions.
When initial testing has been completed, the actual experiment can proceed. Cells can be batch sorted with hundreds to thousands of cells per tube or individual cells can be sorted into 96-well plates for clone isolation. If an inadequate level of enrichment is achieved after the first round of sorting, cells can be cultured and then resorted. This process can be repeated until the desired level of enrichment is achieved, and final processing of the cells will be highly dependent on experimental design. Cells can be further cultured for DNA isolation, or PCR amplification of genetic elements can be performed from bacterial cultures and/or single cells. Other protocols may culture sorted cells before isolating proteins for various in vitro assays.
VII. CONCLUSIONS
The sections presented herein serve to provide the sorter with a foundation in appropriate sorting techniques, as well as to provoke thought toward elegant experimental design. These approaches take advantage of the latest advances in molecular biology as well as the full range of capabilities inherent to high-end cell sorters. As sorting technology has improved, these instruments have become more stable, easier to operate, and amenable to almost any laboratory setting as opposed to dedicated, core-sorting facilities. The highspeed cell sorter has become a tool for discovery -diversity of heterogeneous populations can be assessed, entire genomes and molecular libraries can be screened in short periods of time, pure populations can be purified, and their constituents can be amplified and studied further with minimal confounding variables. No other technology has the ability to purify cells based on so many parameters with such high speed and accuracy as the high-speed cell sorter. The tight interplay between biology and technology has resulted in an improved ability to highlight increasingly specified nuances between cells. In combination w i t h the latest tools, such as mass spectrometry and expression arrays, these differences will unravel the earliest molecular underpinnings in processes such as disease pathogenesis and develop - mental biology.
References
Barker, L. P., Brooks, D. M., and Small, P. L. (1998). The identification of Mycobacterium marinum genes differentially expressed in macrophage phagosomes using promoter fusions to green fluorescent protein. Mol. Microbiol. 5, 1167-1177.
Boder, E. T., Midelfort, K. S., and Wittrup, K. D. (2000). Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. LISA 97(20), 10701-10705.
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263(5148), 802-805.
Chong, S. S., Almqvist, E., et al. (1997). Contribution of DNA sequence and CAG size to mutation frequencies of intermediate alleles for Huntington disease: Evidence from single sperm analyses. Hum. Mol. Gene. 6, 301-309.
Gray, J. W., Dean, P. N., Fuscoe, J. C., Peters, D. C., Trask, B. J., van den Engh, G. J., and Van Dilla, M. A. (1987). High-speed chromosome sorting. Science 238(4825), 323-329.
Inouye, S., Ogawa, H., et al. (1997). A bacterial cloning vector using a mutated Aequorea green fluorescent protein as an indicator. Gene 189, 159-162.
Kalakonda, N., Rothwell, D. G., et al. (2001). Detection of N-Ras codon 61 mutations in subpopulations of tumor cells in multiple myeloma at presentation. Blood 98, 1555-1560.
Koo, J. T., Choe, J., and Moseley, S. L. (2004). HrpA, a DEAH-box RNA helicase, is involved in mRNA processing of a fimbrial operon in Escherichia coli. Mol. Microbio.
Matz, M. V., Fradkov, A. E, Labas, Y. A., Savitsky, A. P., Zaraisky, A. G., Markelov, M. L., and Lukyanov, S. A. (1999). Fluorescent proteins from nonbioluminescent Anthozoa species. Nature Biotechnol. 10, 969-973.
Olsen, M. J., Stephens, D., Griffiths, et al. (2000). Function-based isolation of novel enzymes from a large library. Nature Biotechnol. 18, 1071-1074.
Peters, D., Branscomb, E., Dean, P., Merrill, T., Pinkel, D., Van Dilla, M., and Gray, J. W. (1985). The LLNL high-speed sorter: Design features, operational characteristics, and biological utility. Cytometry 6(4), 290-301.
van den Engh, G. (2000). High speed cell sorting. In "Emerging Tools for Single Cell Analysis: Advances in Optical Measurement Technologies" (G. Durack, and J. P. Robinson, eds.). Wiley-Liss, New York.
Van Dilla, M. A., and Deaven, L. L. (1990). Construction of gene libraries for each human chromosome. Cytometry 11(1), 208-218.
Support our developers




