Human Genome Project Resources for Breakpoint Mapping
I. INTRODUCTIONThe announcement in April 2003 that the Human Genome Project was complete and was available publicly through intuitive genome browsers (e.g., see http://www.ensembl.org/Homo_sapiens/ and http://genome.ucsc.edu/cgi-bin/hgGateway) revolutionised experimental genome analysis. More precisely the study of chromosome rearrangements has been facilitated by the availability of easily accessible, ordered, fully sequenced tiling path clones covering the entire human genome. This resource enables chromosome breakpoints to be mapped more rapidly and with increased confidence.
Conventionally a chromosome rearrangement has been mapped by walking along a chromosome sequentially hybridising fluorescently labelled DNA clones (FISH) until the signal is no longer retained on one derivative chromosome but is seen on the reciprocal derivative chromosome. The genomic position of the breakpoint is then delineated by these two clones and the spanning clone can be found by FISHing clones at a higher resolution (McMullan et al., 2002). The genome annotation also now easily available on web browsers (e.g., http://vega.sanger.ac.uk/ Homo_sapiens/) provides an additional level of information regarding the gene content of a genomic region of interest. Both known and predicted genes are detailed along with additional information, including repeat content, GC content, mouse homology, and many additional features. This information, together with the identification of breakpoint spanning clones linked directly to these databases, allows candidate genes involved in the phenotype of a patient to be identified readily.
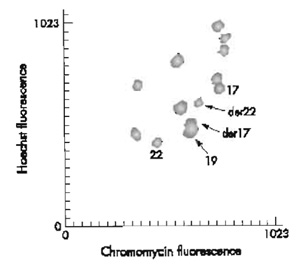 |
| FIGURE 1 Partial flow karyotype of t(17;22)(q21.1;q12.2). |
Clones sequenced as part of the Human Genome Project are available readily to international researchers for minimal cost (e.g., BACPAC resources: http://bacpac.chori.org/, Invitrogen: http://clones. invitrogen.com/, and the Resources for Molecular Cytogenetics, University of Bari: http://www.biologia. uniba.it/rmc/). The sequence obtained from these clones is freely accessible (e.g., http://www.ncbi.nlm. nih.gov/genome/clone/, http://www.ensembl.org/ Homo_sapiens/, and http://genome.ucsc.edu/cgi-bin/hgGateway).
The availability of such a comprehensive set of clones has not only greatly facilitated conventional approaches to breakpoint mapping studies but has also enabled the development of genome-wide, large insert clone DNA microarrays. The selection, DNA extraction, and labelling of clones for multiple sequential DNA clone in situ hybridisation experiments required to map breakpoints are often time-consuming processes. However, the clones can be used as ordered targets themselves for breakpoint mapping after gridded assembly on a DNA microarray (Fiegler et al., 2003a). DNA microarrays have been recently developed mostly with the aim of studying genome imbalance in tumour DNA samples (Solinas-Toldo et al., 1997). We have exploited this resource to study structural chromosome rearrangements, including rearrangements with no genomic imbalance. This type of analysis, termed array painting (Fiegler et al., 2003b), involves isolating derivative chromosomes away from other chromosome homologues by flow sorting (Carter, 1994) or microdissection prior to hybridisation onto the DNA microarrays.
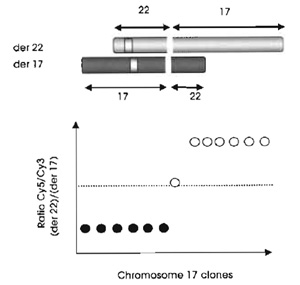 |
| FIGURE 2 Basic principls of mapping translocation breakpoints by array painting. |
The limit of array painting as a tool for mapping chromosome breakpoints is defined by the resolution of the clones gridded. At present we use a 1-Mb array to define the breakpoints to within a megabase (see Fig. 3) and then use conventional FISH methods (see article by Leversha) to refine the breakpoints and find the spanning clones. Clones that span the breakpoint show intermediate ratios on the array (see Fig. 2). Statistically, 1 in every 10 breakpoints will have spanning clones identified directly on the 1-Mb resolution array.
This technique has proven to be invaluable in the detailed study of chromosome rearrangements. When used in conjunction with array CGH, it can give an insight into rearrangements that might have been undetected by G-banding analysis. Increased array resolution, e.g., a tiling path array, in the study of simple chromosome rearrangements would identify spanning clones within a single experiment.
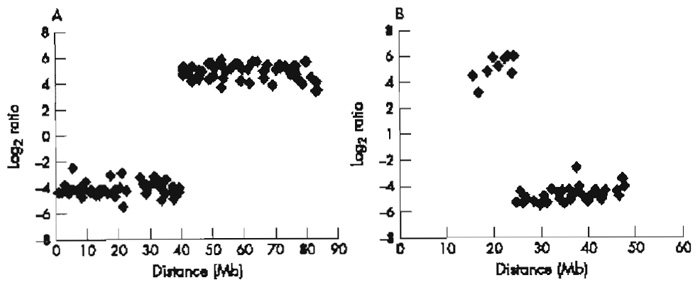 |
| FIGURE 3 Array painting profiles for the derivative chromosomes of t(17;22): (A)1-Mb array chromosome 17 plot and (B)1-Mb array chromosome 22 plot. |
II. MATERIALS AND INSTRUMENTATION
A. Purification of Flow-Sorted DNA
N-Lauroylsarcosine sodium salt (Sigma, L5125)
Proteinase K (Invitrogen, 25530-015)
Phenylmethylsulfonyl fluoride (PMSF) (Sigma, P7626)
Pellet Paint nonfluorescent coprecipitant (Novagen, 70748)
B. Random Labeling of DNA for Array Painting
BioPrime DNA labeling kit (Invitrogen, 18094-011) contains 2.5× random primers solution, sterile water, Klenow fragment, and stop buffer
dNTPs (Amersham Biosciences, 27-2035-02)
Cyanine 3-dCTP (Perkin Elmer, NEL576001EA)
Cyanine 5-dCTP (Perkin Elmer, NEL577001EA)
Microspin G50 columns (Amersham Biosciences, 27- 5330-01)
C. Array Hybridization
Herring sperm DNA (Sigma, D7290)
Human Cot-1 DNA (Roche, 1581074)
Deionised formamide for hybridisation buffer (Sigma, F9037)
Dextran sulphate sodium salt (Amersham Biosciences, 17-0340-02)
HPLC water (VWR International, 152736D)
Rubber cement (Weldtite, 02002)
Formamide (Sigma, 47670)
Yeast tRNA (Invitrogen, 15401-029)
Tween 20 (VWR International, 663684B)
Hellendahl jars (RA Lamb, E95)
Glass troughs (RA Lamb, E106 + E98)
Parafilm (Sigma, P7793)
Shake "n" stack hybridisation oven (Thermo Hybaid, HBMOJCT)
III. PROCEDURES
A. Purification of Flow-Sorted DNA
Solutions
- 0.25 M EDTA/10% sodium lauroyl sarcosine: For 10 ml, dissolve 1 g N-lauroylsarcosine in 5 ml filtered water and add 5ml 0.5M EDTA (pH 8.0). Store in 1ml aliquots at 4°C.
- Profeinase K: Resuspend 100mg in 5 ml sterile water (final concentration of 20mg/ml). Store 50µl aliquots at -20°C. Do not freeze thaw.
- PMSF: Resuspend 250mg in 12.5ml 96% ethanol (final concentration of 20mg/ml). Store at 4°C up to a maximum of 6 months.
- 5M NaCl: Dissolve 29.22g NaCl (MW 58.44) in 100ml sterile water. Autoclave and store at room temperature.
- Pellet Paint: Store as 20µl aliquots at -20°C. Avoid repeated freeze thawing. Working tube can be kept at 4°C.
- Absolute ethanol: Store in a sterile 50ml Falcon tube at -20°C.
- 70% ethanol: Store in a sterile 50ml Falcon tube at -20°C.
- Sterile T1.0E (pH 8.0): For 100ml, mix 1ml 1M Tris-HCl (pH 8.0), 1ml 0.1M EDTA (pH 8.0), and 98 ml water. Autoclave and store at room temperature.
Steps
Handle tubes containing flow-sorted material very gently!
Flow-sorted chromosomes come in batches of approximately 250,000 per 1.5ml microfuge tube, sorted into chromosome sheath buffer [10mM Tris-HCl (pH 8.0), 1mM EDTA, 100mM NaCl, 0.5 mM Na azide]. Sort 250,000 chromosomes into a volume of approximately 300µl. Amounts of reagents will vary according to the quantity of sorted sample.
- Add 30µl 0.25M EDTA/10% sodium lauroyl sarcosine and 3 µl proteinase K. Incubate overnight at 42°C.
- Dilute stock PMSF to 4mg/ml in 96% ethanol. Add 3.33 µl to sorted DNA tubes (40 µg/ml final). Incubate for 40min at room temperature.
- Add 13.44 µl 5M NaCl (final 0.3M including sheath buffer), 2µl Pellet Paint, and 770µl absolute ethanol. Mix gently by inversion. Precipitate overnight at -20°C or at -70°C for 30min. (Tubes can be stored at this stage at -20°C if necessary.)
- Pellet DNA in microfuge at full speed (11,000g) at room temperature for 15min. The hinge of the Eppendorf tube should face outwards so that the pellet is easily located.
- Remove most of the supernatant with a P1000. Remove the remainder with P200, avoiding the pellet. Add 1ml 70% ethanol without disturbing the pellet and then spin again for 7 min. Remove supernatant as before. Allow pellets to air dry (about 20min).
- Add 50µl TE directly to the pellet to resuspend. DNA should separate from the wall of the tube immediately. Mix by very gentle flicking. Allow DNA to dissolve for at least 2h.
B. Random Labeling of DNA for Array Palnting Solutions
- 2.5× random primer solution: Found in BioPrime kit. Store at -20°C and use as supplied.
- Sterile water: Found in BioPrime kit. Store at -20°C and use as supplied.
- 10× dNTP mix: To make 1ml, mix 10µl dCTP (1 mM final), 20 µl dATP (2 mM final), 20 µl dGTP (2 mM final), and 20µl dTTP (2mM final) in 930µl TE buffer. Store 1ml aliquots at -20°C.
- Cy3-dCTP: Store as stock in the dark at -20°C.
- Cy5-dCTP: Store as stock in the dark at -20°C.
- Klenow fragment: Found in BioPrime kit. Store at -20°C and use as supplied.
- Stop buffer: Found in BioPrime kit. Store at -20°C and use as supplied.
-
Tubes containing Cy dyes should be kept in the dark!
Steps
1. Labeling Reactions
- To 50µl resuspended chromosomes, add 60µl 2.5× random primers solution and add 20.5µl sterile water to make up to 130.5µl.
- Denature samples in a hot block for 10min at 100°C and immediately cool on ice.
- Add the following reagents on ice: 15µl 10x dNTP mix, 1.5µl Cy3- or Cy5-1abelled dCTP, and 3µl Klenow fragment. Mix thoroughly but gently.
- Incubate the reaction at 37°C overnight.
- Stop the reaction by adding 15µl stop buffer.
2. Clean-up of Labeling Reactions
Use 3 columns per labelling reaction. Once the columns have been prepared, they must be used immediately to avoid the resin drying out.
Column Preparation
Steps
- Resuspend the resin in the columns by gentle vortexing.
- Loosen the cap one quarter turn and snap off the bottom closure.
- Place the column in a 1.5 ml screw-capped tube.
- Spin the columns for exactly 1 min at 735g.
Sample Application Steps
- Place the columns in a new 1.5 ml tube and slowly apply 55µl of the labelled sample to the centre of the angled surface of the resin bed, being careful not to disturb it. Do not allow any of the sample to flow around the side of the resin bed.
- Spin the columns for 2 min at 735g. Collect the purified samples in the support tube.
- Discard the columns.
- Combine the three samples.
- Run 5µl on a 1% agarose gel to check for correct labelling. Ideally, the fragments should range from 200 to 800bp in size. Tubes containing the samples should be pink (Cy3) and blue (Cy5) in colour, indicating incorporation of the fluorescent dCTPs.
C. Array Hybridization
Solutions
- Human Cot-1 DNA: Use as supplied (1 µg/ul). Store at -20°C.
- 3 M NaAc pH 5.2: For 200ml, dissolve 49.2 g NaAc (anhydrous) in ~140ml H2O, pH to 5.2 with glacial acetic acid, make up to 200ml, and then autoclave. Store at room temperature.
- Absolute ethanol: Store in a sterile 50 ml Falcon tube at -20°C.
- Herring sperm DNA: Use as supplied (10mg/ml). Store at -20°C.
- 40%formamide/2xSSC: To make up 50ml, mix 20ml formamide, 5 ml 2x SSC, and 25 ml sterile double distilled water (DDW). Store in a 50 ml Falcon at 4°C.
- Hybridisation buffer: For 10ml, mix 5ml deionised formamide, 2 ml 50% dextran sulphate, 100 µl Tris, pH 7.4, 10µl Tween 20, 1ml 2x SSC, and 1890µl HPLC water.
- 80% ethanol: Store in a sterile 50ml Falcon tube at -20°C.
- Yeast tRNA: Make up to a final concentration of 100µg/µl by dissolving in autoclaved water and store as 50-µl aliquots at -20°C.
- 20% formamide/2xSSC: To make up 50ml, use 10ml formamide, 5 ml 2x SSC, and 35 ml DDW. Store in a 50ml Falcon at 4°C.
- PBS/O.05% Tween 20: Add 500µl Tween 20 to 1 liter phosphate-buffered saline (PBS).
- 50% formamide/2xSSC: For 400ml, mix 200ml formamide, 40ml 20xSSC, and 160ml HPLC water. Incubate at 42°C prior to use.
1. Precipitation
Steps
- Prepare 2 × 2ml Eppendorfs.
- Heat the herring sperm DNA on a 70°C hot block for 5 min before use.
- To tube 1 add 180µl Cy3-labelled DNA, 180µl Cy5- labelled DNA, 135 µl human Cot-1 DNA, 55 µl 3 M NaAc, pH 5.2, and 1400µl 100% ice-cold ethanol.
- To tube 2 add 80µl herring sperm DNA, 135µl human Cot-1 DNA, 23µl 3M NaAc, pH 5.2, and 600 µl ice-cold 100% ethanol.
- Mix the tubes gently and precipitate in the dark at -20°C overnight or at -70°C for 30min.
2. Preparation of Slide
Steps
- Place the slide over a dummy slide identifying the array area and apply a rubber cement ring closely around the grid using a small syringe, taking care not to go over the grid. Avoid thin threads of the cement going onto the array as you pull the syringe tip away.
- After the first layer has dried, carefully apply a second layer over the first one.
3. Preparation of Humidity Chambers
Steps
- In a fume hood, prepare a humidity chamber by pipetting a 40% formamide/2xSSC mix onto a Whatman paper strip until the paper is damp.
- Leave the humidity chamber in the fume hood ready for use.
4. Prehybridization
Steps
- Preheat the hybridisation buffer in a 70°C hot block.
- Spin the precipitated DNAs for 15 min at 9500g.
- Remove the supernatant and add 500µl 80% ice-cold ethanol.
- Respin at 9500g for 5 min.
- Remove the supernatant with a P1000 and then a P200.
- Respin at 9500g for 1 min.
- Remove any remaining supernatant with a P10 until the pellet is dry.
- Resuspend the DNAs in 60µl hybridisation buffer and 6µl yeast tRNA (tube 1) and 140µl hybridisation buffer (tube 2). This is best done by adding the hybridisation buffer and leaving the tube for 2-3 min in a 70°C hot block before resuspending the pellet using a displacement pipette. Ensure that the DNA is properly resuspended before continuing with the experiment. Denature the tube for 10min at 70°C. Mix the sample after 5 min.
- Pulse spin the tubes.
- Place tube 1 into a 37°C hot block and incubate for 60min in the dark.
- Apply the contents of tube 2 onto the array grid and rock the slide to ensure even coverage within the well. Remove any air bubbles with a tip, but do not touch the grid. Work quickly to minimise drying of the slide.
- Transfer the slide into the humidity chamber and place on the rocking table for 60min shaking at 5 rpm.
5. Hybridization
Steps
- Pulse spin tube 1.
- Remove slide from humidity chamber.
- Apply DNA onto the slide. Remove any air bubbles.
- Rock the slide to mix the prehybridisation and hybridisation solutions and to ensure even coverage within the well.
- Place the slide into a slide mailer humidified with 20% formamide/2xSSC.
- Seal well with Parafilm and place into the hybridisation oven at 37°C.
- Incubate with gentle rocking (5rpm) for 48h, turning the slide by 90° after 24h.
6. Washing
Steps
- Remove the slide from the incubator and increase the temperature of the hybridisation oven up to 42°C.
- Remove the rubber cement.
- Place the slide into a hellendahl jar containing PBS/0.05% Tween 20 to wash off excess hybridisation solution.
- Transfer the slide into a glass trough and wash in PBS/0.05% Tween 20 for 10 min shaking at room temperature.
- Transfer the slide into a preheated trough containing 50% formamide/2x SSC at 42°C. Incubate at 42°C with shaking for 30 min.
- Transfer the slide into a trough of fresh PBS/0.05% Tween 20 and incubate at room temperature with shaking.
- Transfer the slide into a metal rack and spin at 150g for 5min to dry.
- Store the slide in a light-proof box at room temperature until ready to scan.
7. Scanning and Analysis
Our slides are scanned using an Axon 4000B scanner (Axon Instruments, Burlingame, CA) and the images are analysed by GenePix Pro 3.0 software (Axon Instruments, Burlingame, CA). Spots are defined using the automatic grid feature and adjusted manually where necessary. Fluorescence intensities of all spots are calculated after local background subtraction. The ratios of the intensities are then plotted in log2 scale against the distance of the clones along the chromosomes from the p termini. Breakpoints are identified by a shift in ratio intensities typically from 4 log 2 to-4 log 2 (or vice versa), with a spanning clone showing an intermediate ratio close to 0 log 2.
IV. PITFALLS
Batches of Cot-1 DNA need to be tested before being used routinely in experiments. At present we order Cot-1 DNA from Roche (1581074) or Invitrogen (15279- 011) and perform test hybridisations on the array using tumour cell line DNA with previously determined characteristic genomic copy number changes.
During array hybridisation, care must be taken to prevent the slides from drying out. Just before the hybridisation solution is added to the slide, some of the pre-hyb solution can be removed carefully with a pipette tip. However, this increases the chances of the slide drying out.
All gels used in this article are 1% agarose gels with ethidium bromide. It is advised that these gels are run before progression to the next step to check that each process has been performed successfully.
References
Carter, N. R (1994). Bivariaee chromosome analysis using a commercial flow cytometer. Methods Mol. Biol. 29, 187-204.
Fiegler, H., Carr, R, Douglas, E. J., Burford, D. C., Hunt, S., Smith, J., Vetrie, D., Gorman, R, Tomlinson, I. R M., and Carter, N. R (2003a). DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer 36, 361-374.
Fiegler, H., Gribble, S. M., Burford, D. C., Carr, R, Prigmore, E., Porter, K. M., Clegg, S., Crolla, J. A., Dennis, N. R., Jacobs, R, and Carter, N. P. (2003b). Array painting: A method used for the rapid analysis of aberrant chromosomes using DNA microarrays. J. Med. Genet. 40, 664-670.
McMullan, T. E W., Crolla, J. A., Gregory, S. G., Carter, N. R, Cooper, R. A., Howell, G. R., and Robinson, D. O. (2002). A candidate gene for congenital bilateral isolated ptosis identified by molecular analysis of a de novo balanced eranslocation. Hum. Genet. 110, 244-250.
Solinas-Toldo, S., Lampei, S., Stilgenbauer, S., Nickolenko, J., Benner, A., Döhner, H., Cremer, T., and Licheer, P. (1997). Matrix-based comparative genomic hybridization: Biochips to screen for genomic imbalances. Genes Chromosomes Cancer 20, 399-407.
Telenius, H., Carter, N. P., Bebb, C. E., Nordenskj61d, M., Ponder, B. A. J., and Tunnacliffe, A. (1992). Degenerate oligonucleoeideprimed PCR: General amplification of target DNA by a single degenerate primer. Genomics 13, 718-725.
Support our developers




