Immunofluorescence Microscopy of the Cytoskeleton: Combination with Green Fluorescent Protein Tags
I. INTRODUCTIONImmunofluorescence microscopy is now a standard procedure for the localisation of molecules in cells (for an introduction to the method, see article by Osborn). Nevertheless, the method has its pitfalls, not least in requiring the immobilisation of cells by chemical fixation and multiple manipulations during labelling. This can lead to the loss of antigen as well as to the distortion of cell structure. Unfortunately, published pictures still appear in which distortions in cell structure are overlooked and where the conclusions drawn are consequently tenuous. Advances in live cell imaging, combined with green fluorescent protein (GFP) tags (and analogues) now provide independent methods for assessing the localisation of molecules. Nevertheless, immunofluorescence microscopy remains an important technique when used with discretion. Indeed, when applied in combination with cells expressing fluorescently tagged probes, it adds a further dimension to characterise the relative localisation of multiple components. This article provides some recipes suitable for labeling the cytoskeleton and gives examples where GFP tags and immunofluorescence can be usefully combined.
II. MATERIALS AND REAGENTS
- Coverslips: Round glass coverslips 12 or 15 mm in diameter, cleaned in 60% ethanol/40% HCl (10 min), rinsed with H2O (2 × 5min), drained, cleaned with lint-free paper, and sterilized for tissue culture by exposure to ultraviolet light in the culture dish.
- Humid chamber: Large petri dish 14 cm in diameter, or similar container with lid, containing a glass plate (around 9cm2) coated with a layer of Parafilm and supported on a moistened piece of filter paper on the bottom of the dish. A few drops of water on the glass plate facilitate spreading and flattening of the Parafilm.
- Washing reservoir: Two multiwell dishes, 24 wells each (e.g., Nunc).
- Filter paper: Whatman No. 1, 9 cm in diameter.
- Forceps: Dumont No. 4 or No. 5 or equivalent watchmaker forceps.
- Pipettes: Set of automatic pipettes (0-20, 20-200, 50-1000µl) or capillary pipettes for diluting and aliquoting antibodies. Pasteur pipettes.
- Phalloidin: Alexa 568- and Alexa 488-coupled phalloidins from Molecular Probes and CPITC (coumarin) phalloidin from Sigma. Store as 0.1-mg/ml stocks in methanol at -20°C.
- Secondary antibodies: Commercial secondary antibodies carrying Molecular Probes Alexa 488, 568, and 350 conjugates are, in our experience, of generally good quality.
- Gelvatol, Vinol: The basic ingredient of the mounting medium is polyvinyl alcohol (MW 10.000, around 87% hydrolysed), which comes under various trade names: Elvanol, Mowiol, and Gelvatol. We use Vinol 203 from Air Products and Chemical Inc.
II. PROCEDURES
Solutions
- 0.5M EGTA stock solution: For 500 ml stock, weigh out 95.1 g EGTA (Sigma E-4378) into 400ml H2O, adjust pH to 7.0 with 1 N NaOH, and make up to 500 ml with H2O. Store at room temperature in a plastic bottle.
- 1M MgCl2 stock solution: For 500ml stock, weigh out 101.6g MgCl2, add H2O to 500ml, dissolve, and store at 4°C.
- Cytoskeleton buffer (CB): 10mM MES (Sigma M- 8250), 150 mM NaCl, 5 mM EGTA, 5 mM MgCl2, and 5 mM glucose. For 1 liter, add the following amounts to 800 ml H2O: MES, 1.95 g; NaCL, 8.76 g; 0.5 M EGTA, 10 ml; 1M MgCl2, 5 ml; and glucose, 0.9 g. Adjust pH to 6.1 with 1N NaOH and fill up to 1 liter. Store at 4°C. For extended storage, add 100 mg streptomycin sulfate (Sigma S-6501).
- Phosphate-buffered saline (PBS) working solution: 137mM NaCl, 2.7mM KCl, 4.3mM Na2HPO4·7H2O, and 1.4mM KH2PO4, pH 7.4.
- Triton X-100: Make up 10% aqueous stock and store at 4°C.
- Glutaraldehyde (GA) stock: Make up 2.5% solution of glutaraldehyde by diluting 25% glutaraldehyde EM grade (Agar scientific Ltd., Cat. No. R 1020 or equivalent) in CB. Readjust pH to 6.1 and store at 4°C.
- Paraformaldehyde (PFA) stock: Make up a stock 4% solution of paraformaldehyde in PBS or CB (see fixative mixtures) (analytical grade Merck Cat. No. 4005). To make 100ml, heat 80ml of CB (or PBS) to 60°C, add 3g paraformaldehyde, and mix 30min. Add a few drops of 10M NaOH until the solution is clear, cool, adjust pH (see appropriate mixture), and make up to 100ml. Store in aliquots at -20°C.
- Fixative mixtures: Aldehyde fixative mixtures are made up using the stock solutions given earlier to give the combinations listed under step 2.
- Blocking solution: 1% bovine serum albumin and 5% horse serum in PBS.
- Antibody mixtures: These are made up in PBS or in the blocking solution without serum. To remove any unwanted particles, centrifuge (10,000 g for 10 min) the diluted mixture before use. The antibody combinations used for this article are listed in Table I.
- Mounting medium: Mix 2.4 g of polyvinyl alcohol with 6g glycerol (87%) and then with 6ml H2O. After at least 2h at room temperature, add 0.2ml 0.2M Tris-HCl, pH 8.5, to the mixture and further incubate the solution for 10min at 60°C. Remove any precipitate by centrifugation at 17,000g for 30min. Store in aliquots at -20°C. [Antibleach agents are available that considerably reduce bleaching and thus enable multiple pictures to be taken of the same cells. We use n-propyl gallate (Giloh and Sedat, 1982) at 5mg/ml or phenylenediamine (Johnson et al., 1982) at 1-2mg/ml in the mounting medium. After dissolving the additive, degas mounting medium before storage.]
Steps
- Seed the cells onto coverslips in the petri dish and allow them to attach and spread for 4-48 h in an incubator at 37°C.
- Aspirate growth medium and rinse dish gently
with PBS (warmed 37°C PBS); avoid shifting of the
coverslips over each other. Aspirate PBS and replace
with one of the following fixative solutions.
- Fix 1: 4% PFA/0.1% Triton X-100 in PBS for 2min. Rinse three times with PBS. 4% PFA in PBS for 20 min. Wash 2 × 10 min with PBS.
- Fix 2: 0.5% GA/0.25% Triton X-100 in CB for 1 min. Rinse 3× with CB. 4% PFA in CB for 20 min. Wash 2 × 10 min with CB.
- Fix 3: 0.25% GA/0.5% Triton X-100 in CB for 1 min. Rinse 3× with CB. 1% GA for 15 min. Wash 2 × 10 min with CB.
- Fix 4: 4% PFA/0.3% Triton X 100/0.1% GA in CB for 15 min. Wash 2 × 10min with CB.
- Block: Invert each PBS coverslip onto a 30-µl drop of blocking solution on Parafilm in the humid chamber. Before transfer to drop, dry the back side of the coverslip by holding it briefly on filter paper with a pair of forceps, taking care not to allow the cell side to dry. Drain any excess solution from the cell side by touching the edge of the coverslip to the filter paper. Incubate on blocking solution for 15 min or until first antibody mixtures are prepared. (Back side of coverslip should not be wet or else coverslip will sink during the washing step.)
- Apply drops (20-50µl) of first antibody mixture to unused part of Parafilm and transfer coverslips to appropriate drops after draining excess blocking solution on filter paper. Replace lid on petri dish and leave at room temperature for 45-60 min.
- Wash: To ease removal of coverslips for washing, pipette 100 µl PBS under their edge to lift them up from the Parafilm. Using forceps, transfer coverslips to a multiwell dish in which the wells are filled to the brim with PBS so that the liquid surface is fiat. The coverslips will float well, cell side down, as long as the back side remains dry. (For efficient washing, transfer dish gently to a tilting rotating table for 10min.) Repeat washing steps after transfer of coverslips to a second dish containing fresh PBS two or three times.
- Change Parafilm in humid chamber and apply drops of second antibody mixture. Transfer coverslips to drops after briefly draining excess with filter paper and incubate for 45 min.
- Wash as described in step 6.
- Mount: Add a small drop of mounting medium to a cleaned glass slide using, for example, a plastic disposable pipette tip. Drain excess (PBS) from coverslip and gently invert onto drop. Note: The mounting medium dries quite fast so the drops should be applied singly and not in batches. If necessary, remove excess medium after mounting by applying small pieces of torn filter paper to the coverslip edge.
- Observe directly in a fluorescence microscope with a dry lens. An oil immersion lens can be used the next day when the mounting medium has solidified. Alternatively, the drying time can be shortened by transfer of slides to a 27°C oven.
III. CHOICE OF FIXATION FOR MULTIPLE LABELLING
Different antibodies commonly require different fixation protocols to give optimal labelling. It is therefore important to test different fixation conditions for each antibody to determine the best compromise fixation for multiple labelling. Table I lists the characterics of the commercial antibodies used in this article in terms of the intensity of label obtained with each of the four fixation protocols described for cultured smooth muscle cells (A7r5, American Type Culture Collection). In general, you should establish the strongest fixation protocol that your antibodies can tolerate and draw your conclusions accordingly.
 |
IV. COMBINING IMMUNOFLUORESCENCE AND GFP TAGS
The ability to express fluorescent proteins as tags to gene products in living cells represents an important advance in localisation methods. In addition to facilitating the visualisation of protein dynamics in vivo with sensitive imaging systems, these tagging methods can be usefully combined with the imunofluorescence procedure. Additional flexibility is offered by the fact that the tagged protein is already fluorescent so that restrictions with the use of secondary antibodies are reduced and triple labelling is relatively straightforward. With the cytoskeleton, phalloidin is a probe of choice for actin; when applying this probe, four labels in one cell would not be problematic, given a suitable choice of fluophores (including Cy-5 in the infrared, for example).
An interesting aspect of proteins that are washed away easily during fixation is that they are retained more readily when tagged with a GFP moiety (M. Gimona, unpublished observations). This property offers an unexpected advantage of GFP-tagged probes for localisation studies with fixed cells.
Figures 1-3 show examples of cells expressing GFPtagged proteins and then fixed and labelled with phalloidin (for actin) and a single antibody. The fixation procedure was "fixation 1" in step 2.
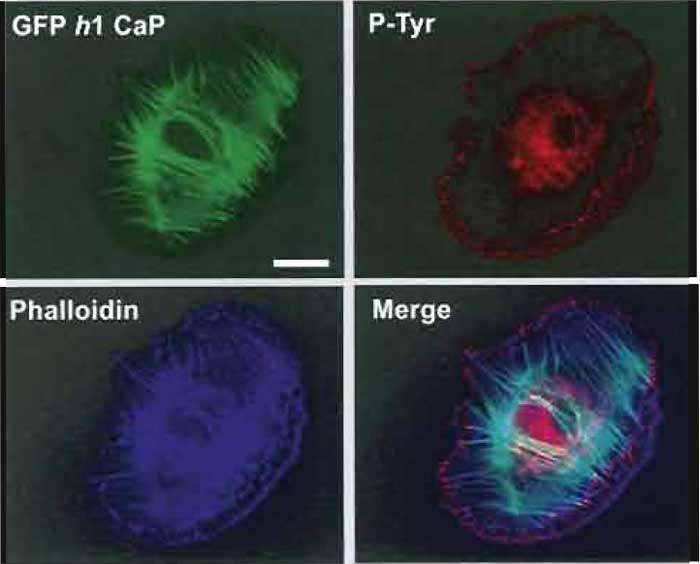 |
| FIGURE 1 Flourescence images of an A7r5 smooth muscle cell transfected with GFP h1 calponin (Gimona, 2003) and then counterstained with Alexa phalloidin 350 (dilution, 1:200) and mouse antiphosphotyrosine (PY99 Santa Cruz dilution 1 : 1000), followed by a GaM Alexa 568 as secondary antibody at a dilution of 1:750. Images were recorded on a Zeiss Axioskop fitted with an Zeiss Axiocam imaging system. All three images are combined in the merged image (bottom right). Bar: 20µm. |
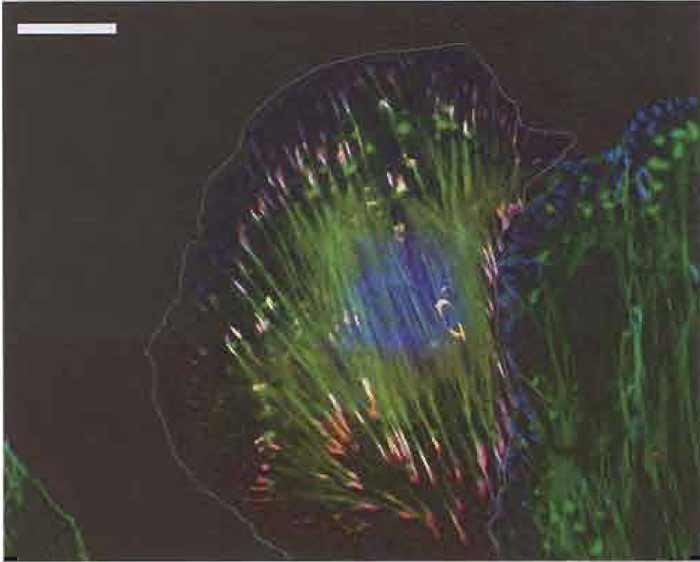 |
| FIGURE 2 Merged fluorescence images (as Fig. 1) of an A7r5 smooth muscle cell transfected with Ds-Red zyxin (Bhatt et al., 2002) to mark focal adhesions and then labelled with Alexa 488 phalloidin (dilution, 1:300) and antiphosphotyrosine (see Fig. 1) with GaM Alexa 350 at a dilution of 500 as secondary antibody. The boundary of the transfected cell is indicated by a white line. Bar: 20µm. |
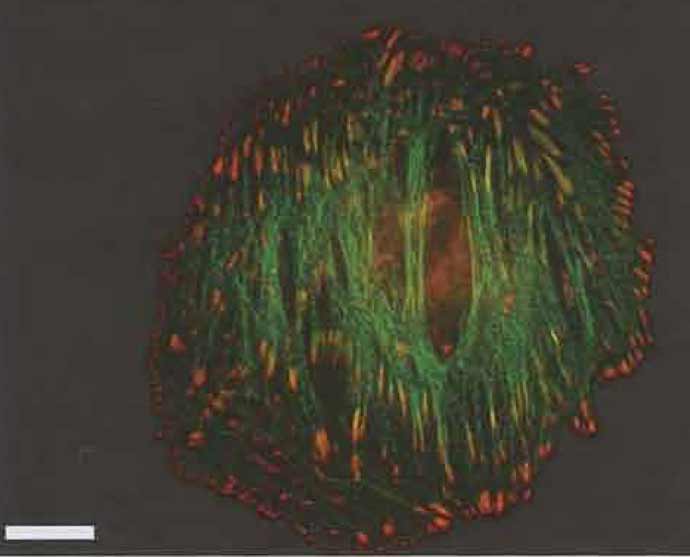 |
| FIGURE 3 Merged fluorescence images (as Fig. 1) of an A7r5 smooth muscle cell transfected with GFP-α-actinin (Gimona et al., 2003) and counterstained with mouse antiphosphotyrosine (as in Fig. 1). Bar: 20 µm. |
V. COMMENTS
We have generally aimed for fixation protocols that best preserve the actin cytoskeleton. Although stress fibers are easily preserved with most fixative protocols, the delicate peripheral lamellipodia are normally distorted or lost after methanol or formaldehyde fixation. This is why glutaraldehyde is included in two of the present mixtures. A stronger glutaraldehyde fixation than what we have used here is best for lamellipodia (see, e.g., Small, 1988) but cannot be used with several of the antibodies described. So again we have had to compromise. Weber and colleagues (1978) introduced the use of sodium borohydride to reduce free aldehyde groups after glutaraldehyde fixation and thereby the autofluorescence introduced by this fixative. If autofluorescence is observed, for example, in the region of the nucleus, this can be eliminated by a brief treatment (3 × 10min) of the coverslips in icecold cytoskeleton buffer containing freshly dissolved sodium borohydride (0.5mg/ml). The coverslips are rinsed three times prior to immunolabelling.
VI. PITFALLS
If problems arise from sinking of coverslips during washing, use another washing protocol, e.g., immersion of coverslips cell side up in separate petri dishes containing PBS. Damaged cells normally arise from inadvertently allowing the coverslip to dry at any stage of the procedure or by touching the cell side with filter paper. Labelling with phalloidin can be improved by including this probe also in the first antibody. Successful double or triple immunofluorescence labelling requires that the individual antibody combinations each produce intense staining with a clean background, when used alone.
References
Bhatt, A., Kaverina, I., Otey, C., and Huttenlocher, A. (2002). Regulation of focal complex composition and disassembly by the calcium-dependent protease calpain. J. Cell Sci. 115, 3415-3425.
Giloh, H., and Sedat, J. W. (1982). Fluorescence microscopy: Reduced photobleaching of rhodamine and fluorescein protein conjugates by n-propyl gallate. Science 217, 1252-1255.
Gimona, M., Kaverina, I., Resch, G. P., Vignal, E., and Burgstaller, G. (2003). Calponin repeats regulate actin filament stabilty and formation of podosomes in A7r5 smooth muscle cells. Mol. Biol. Cell.
Johnson, G. D., Davidson, R. S., McNamee, K. C., Russell, G., Goodwin, D., and Holborow, E. J. (1982). Fading of immunofluorescence during microscopy: A study of the phenomenon and its remedy. J. Immunol. Methods. 55, 231-242.
Small, J. V. (1988). The actin cytoskeleton. Electron Microsc. Rev. 1, 155-174.
Weber, K., Rathke, P. C., and Osborn, M. (1978). Cytoplasmic microtubular images in glutaraldehyde-fixed tissue culture cells by electron microscopy and by immunofluorescence microscopy. Proc. Natl. Acad. Sci. USA 75, 1820-1824.
Support our developers




