In vitro Assays for Mitotic Spindle Assembly and Function
I. INTRODUCTIONDuring .the past two decades, cell biology has entered a phase in which technology is so powerful that fundamental questions concerning the morphogenesis and function of cellular organelles can be addressed. One essential and beautiful structure is the mitotic spindle. The work of Lohka and Maller (1985), followed by that of a few other laboratories (Murray and Kirschner, 1989; Sawin and Mitchison, 1991; Shamu and Murray, 1992), opened up a novel approach to studying such a complex and dynamic structure. The idea is not to purify individual spindle components and put them back together hoping that a spindle will assemble, it is rather to open up the cell and prepare g cytoplasmic extract as crude and concentrated as possible to keep the conditions close to the in vivo situation. At first sight, this approach seems uninformative: one merely mimics in vitro what happens in vivo. However, several methods have been developed to manipulate the system, such as the addition of reagents and depletion of proteins. Also the microscopy techniques have evolved and now allow the study of different aspects of spindle formation and function (Desai et al., 1998; Kalab et al., 2002). Thus, this system can be used both to analyze the mechanism of spindle assembly and function and to evaluate the role of individual molecules in the process. More methods are still being developed or improved that will increase the usage and the utility of these extracts even more.
II. MATERIALS AND INSTRUMENTATION
A. Preparation of Xenopus laevis Egg Extracts
Incubator at 16°C, clinical centrifuge, DuPont Sorvall RC-5 centrifuge, HB-4 or HB-6 rotor with rubber adaptors (Sorvall Cat. No. 00363), Beckman ultraclear SW50 tubes (Cat. No. 344057), Sarstedt 13-ml adaptor tubes (Cat. No. 55.518), 1-ml syringes, 18- and 27-gauge needles, glass pasteur pipettes.
Mature female frogs are from African Reptile Park. Pregnant mare serum gonadotropin (Intergonan) is from Intervet. Human chorionic gonadotropin (HCG, Cat. No. CG-10), cytochalasin D (Cat. No. C-8273), EGTA (Cat. No. E-4378), 4.9 M MgCl2 (Cat. No. 104.20), and ATP (Cat. No. A-2383) are from Sigma. CaCl2 (Cat. No. 2383), KCl (Cat. No. 1.04936.1000), NaCl (Cat. No. 1.06404.1000), and L-cysteine (Cat. No. 1.02838.1000) are from Merck. Leupeptin (Cat. No. E18) and pepstatin (Cat. No. E19) are from Chemicon. Aprotinin (Cat. No. 981532) and creatine phosphate (Cat. No. 0621714) are from Roche. HEPES is from Biomol. Sucrose is from USB (Cat. No. 21938).
B. Preparation of DNA Beads
Biotin-21-dUTP (Cat. No. 5201-1) is from Clontech. Biotin-14-dATP (Cat. No. 19524-016) is from Invitrogen. Thio-dCTP (Cat. No. 27-7360-02), thio-dGTP (27- 7370-04), and G-50 gel-filtration (NICK) columns (17-0855-01) are from Amersham Pharmacia Biotech. Restriction enzymes and Klenow (Cat. No. M02125) are from New England Biolabs. The magnetic particle concentrator (MPC) and Kilobase BINDER kit containing Dynabeads M-280 Streptavidin, washing, and binding solutions can be obtained from Dynal. Trizma base (Cat. No. T-1503) is purchased from Sigma, and EDTA (Cat. No. 1.08418.1000) is from Merck.
C. Spindle and Aster Assays
Freshly prepared CSF extract, sperm nuclei (3000/µl)(Murray, 1991), rhodamine-labelled tubulin (2-3mg/ml) (Hyman et al., 1991), 76 × 26-mm microscope slides, 22 × 22-mm coverslips, fluorescence microscope. PIPES (Cat. No. P-6757), Triton X-100 (Cat. No. T-8787), glutaraldehyde (Cat. No. G-5882), NaHB4 (Cat. No. S-9125), GTP (Cat. No. G-8877), and Hoechst dye (bisbenzimide, Cat. No. H33342) are from Sigma. Glycerol (Cat. No. 1.04093.2500) and formaldehyde (Cat. No. 1.04003) are from Merck. Protein A dynabeads (Cat. No. 100.02) are from Dynal. Dimethyl sulfoxide (DMSO, Cat. No. 27,685-5) is from Sigma- Aldrich, and Paclitaxel (taxol equivalent) (Cat. No. P-3456) is from Molecular Probes. For sedimentation and immunofluorescence experiments, corex tubes (15 ml) are equipped with plastic adaptors (home-made, see Evans et al., 1985) to support 12-mm round coverslips. The tubes are centrifuged in an HB-4 rotor containing rubber adaptors, pGEX expression vectors are from Amersham Pharmacia Biotech.
III. PROCEDURES
A. Preparation of X. laevis CSF Egg Extracts
Good protocols detailing the preparation of Xenopus egg extracts and sperm nuclei have already been published (Murray, 1991). We describe here only the preparation of metaphase cytostatic factor-arrested "CSF" extracts as optimized in our laboratory for spindle assembly reactions.
Solutions
- Pregnant mare serum gonadotropin (PMSG): Dissolve in sterile water to a final concentration of 200 units/ml. Store at 4°C.
- Human chorionic gonadotropin (HCG): Dissolve in sterile water to a final concentration of 2000 units/ ml. Store at 4°C.
- MMR: 100mM NaCl, 2mM KCl, 1 mM MgCl2, 2mM CaCl2, 0.1 mM EDTA, 5 mM HEPES, pH 7.8. Prepare a 20X MMR stock solution. Adjust pH with NaOH, autoclave, and store at room temperature. Before use make 1X MMR from the 20X stock solution and adjust pH again if necessary.
- Dejellying solution: 2% L-cysteine, pH 7.8. Prepare in water and adjust pH with NaOH. Make fresh just before use.
- LP: dissolve leupeptin and pepstatin in DMSO to a final concentration of 10mg/ml. Store in 50-µl aliquots at -20°C.
- Aprotinin: Dissolve in sterile water to a final concentration of 10mg/ml. Store in 50-µl aliquots at -20°C.
- Cytochalasin D: Dissolve in DMSO to a final concentration of 10mg/ml. Store in 50-µl aliquots at -20°C.
- 20X XB salts: 2M KCl, 20mM MgCl2, 2mM CaCl2. Filter sterilize and store at 4°C.
- XB: 1X XB salts containing 50mM sucrose and 10mM HEPES. Adjust pH to 7.7 with KOH. Prepare fresh before use.
- CSF-XB: Prepare from XB buffer by adjusting MgCl2 to 2mM and adding 5mM EGTA. To prepare CSF-XB with protease inhibitors (CSF-XB with PI), add LP stock solutions and aprotinin to a final concentration of 10µg/ml.
- Energy Mix: 20X stock contains 150mM creatine phosphate, 20mM ATP, 2mM EGTA, 20mM MgCl2. Store in 100-µl aliquots at -20°C.
Steps
- Inject four to six frogs subcutaneously with 0.5 ml (1000 units) PMSG each using 1-ml syringes and a 27-gauge needle at least 4 days before planning to make an extract. They should be used within 2 weeks after the priming injection. The number of frogs required depends on the quantity and quality of eggs. Five milliliters of eggs (one SW50 tube) gives approximately 1ml of extract.
- Sixteen to 18h before use, inject frogs subcutaneously with 0.25-0.5 ml (500-1000 units) HCG. Place the frogs in individual boxes containing 500ml MMR at 16°C.
- Prepare all solutions before starting collecting the eggs: 2 liters MMR, 500ml XB, 400ml CSF-XB, 100ml CSF-XB with PI, and 500 ml of dejellying solution. Rinse all glassware with distilled water (eggs stick to plastic dishes). Cut off the end of a glass pasteur pipette and fire polish it to make a wide-mouth pipette.
- Collect laid eggs in MMR. Frogs can also be squeezed, which often gives the highest quality eggs. Keep eggs from different frogs in separate batches. Discard batches of eggs containing more than 5% of lysed, ugly, or stringy eggs.
- Wash a few times with MMR to take away skin and other detritus and remove bad eggs. Pour off MMR and add 250ml dejellying solution. When laid, eggs are enveloped in a transparent jelly coat and do not pack closely together. Swirl the beaker frequently and change the dejellying solution. After removal of the jelly coat, eggs pack together. This takes about 5 min. Eggs left for too long in cysteine will lyse.
- Pour off the dejellying solution and add 500ml MMR. Repeat the rinse one more time. After removal of the jelly coat, the eggs become fragile. They lyse easily and can activate if in contact with air. They must always remain immersed in buffer. Remove all badlooking eggs: white and puffy, flattened, or activated ones (darker pole retracted), and those with mottled pigmentation.
- Wash three times with 100-200ml XB.
- Remove as much buffer as possible, keeping all eggs immersed. Wash three times with 100-200ml CSF-XB and finally keep eggs in CSF-XB with PI.
- Transfer eggs using the cut glass Pasteur pipette to SW50 tubes containing 1ml CSF-XB with PI plus 3µl cytochalasin D (30µg/ml). Always immerse the pipette tip in solution before expelling eggs to prevent contact with air. Transfer the SW50 tubes to 13-ml Sarstedt adaptor tubes, which contain 0.5 ml of water to prevent the tubes from collapsing.
- Centrifuge in a clinical centrifuge at 16°C at 600g for 1 minute and then immediately increase the speed to 1200g for an additional 30 s. After centrifugation, remove all excess buffer from the top of the packed eggs. Removal of buffer is critical to obtain a concentrated cytoplasm.
- Place the tubes in an HB-4 or HB-6 rotor containing rubber adaptors. Centrifuge at 10,500rpm for 15 min at 16°C to crush the eggs.
- Place the tubes on ice. A yellow lipid layer is at the top of the tube. Underneath is the cytoplasmic layer, then heavy membranes, and yolk particles at the bottom of the tube. Wipe the sides of the tubes with a tissue before piercing with a 18-gauge needle at the bottom of the cytoplasmic layer. Slowly and carefully aspirate the cytoplasm using a 1-ml syringe with the needle opening facing upward. Remove needle from syringe and carefully expel the cytoplasm into a 1.5-ml tube. Place on ice.
- Add LP and aprotinin to 10µg/ml (1:1000 dilution of stocks) and cytochalasin D to 20µg/ml (1:500 dilution of stock). Add energy mix (1:20 dilution of stock) and mix gently.
Comments on Sperm Nucleus Preparation
Based on Gurdon (1976) and modified by Murray (1991), our only modification is to mash fragments of testes between two frosted (rough) slides before filtering through a cheesecloth mesh.
B. Preparation of DNA Beads
For preparation of DNA beads, linearized couple plasmid DNA and fill in with nucleotides so that one end contains biotinylated bases and the other thionucleotides to inhibit exonuclease activity and the formation of large aggregates of beads. Couple the DNA to 2.8 µm magnetic beads. Incubation in the extract will allow the association of chromatin proteins and the consequent formation of chromatin (Heald et al., 1996).
Solutions
- TE: 10mM Tris, 1 mM EDTA, pH 8. Adjust pH with HCl, autoclave, and store at room temperature.
- Washing and binding solutions: Included with Kilobase BINDER kit.
- Bead buffer: 2M NaCl, 10mM Tris, 1 mM EDTA, pH 7.6. Adjust pH with HCl and store at room temperature.
- Hoechst dye solution: Dissolve bisbenzimide in water to a final concentration of 10mg/ml. Store in the dark at 4°C.
Steps
- Prepare plasmid DNA by Qiagen column purification. While the sequence of the DNA is not important, the plasmid should be more than 5kb to effectively induce chromatin assembly. Cut 50 µg of the DNA with two restriction enzymes that have unique sites in the polylinker to produce one short and one long DNA fragment. One end of the long fragment should terminate in an overhang containing Gs and Cs and the other should contain only As and Ts (e.g., Not1, BamH1). See Fig. 1.
- Ethanol precipitate the DNA and resuspend in 25 µl TE. Quantify recovery by OD260 measurement.
- Prepare fill-in reaction in 70µl containing 1X Klenow buffer, 30 µg DNA, 50 µM nucleotides (biotindATP, biotin-dUTP, thio-dCTP, and thio-dGTP) and 20 units Klenow. Incubate for 2h at 37°C.
- Remove unincorporated nucleotides, following instructions supplied with the Sephadex G-50 gel filtration column (NICK column). The DNA is eluted in a large volume (400µl), but the recovery is better than with spin columns. Quantify recovery by OD260 measurement.
- Prepare coupling mix by combining 400µl biotinylated DNA and 400µl binding solution. Set aside 25 µl of the coupling mix for later evaluation of coupling efficiency.
- Prepare 3µl of streptavidin-conjugated dynabeads for each microgram of DNA recovered, so 120µl for 30µg. Retrieve beads using the MPC (magnet) and wash once with 5 volumes of binding solution (600µl for 120-µl beads). Retrieve the beads and resuspend them in coupling mix containing DNA.
- Incubate bead/coupling mixture for several hours (or overnight) on a rotating wheel at 4°C.
- Retrieve the beads and save the supernatant. Compare the OD260 of the supernatant to the sample taken before coupling to determine the amount of DNA immobilized. Typically two-thirds of the DNA is coupled. Stain 1 µl of beads with 5µg/ml Hoechst dye and observe them by fluorescence microcopy. The beads should appear as very bright dots with no dark patches. If the amount of DNA coupled in the first round does not seem sufficient, incubate the beads a second time with biotinylated DNA.
- Wash beads twice with washing solution and then twice with bead buffer. After the last wash, resuspend the beads in bead buffer so that the final concentration of immobilized DNA is 1µg/5µl of beads. Store at 4°C.
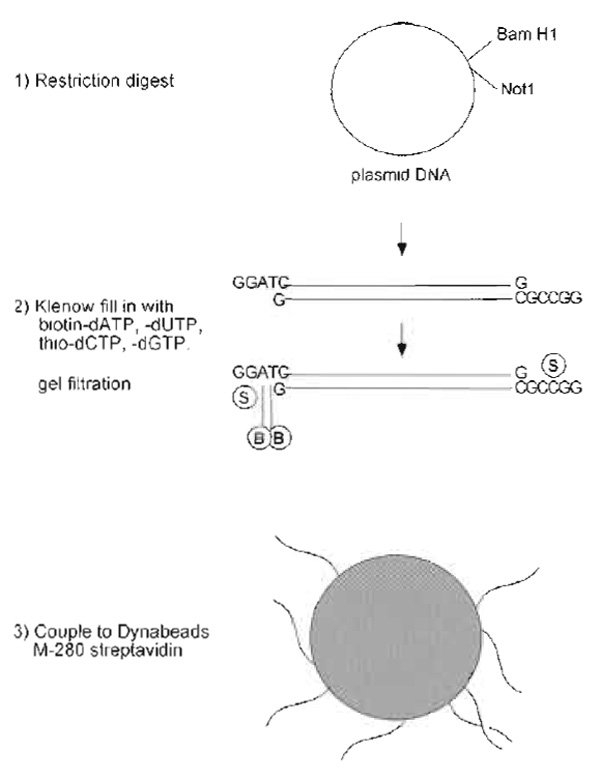 |
| FIGURE 1 Steps in preparation of DNA beads. |
C. Spindle and Aster Assembly in vitro
Chromatin triggers spindle assembly in CSF Xenopus egg extract. Experimentally there are two ways to induce formation of the spindle: by the addition of sperm nuclei (Desai et al., 1999) or DNA beads to the extract (Heald et al., 1996). To decide on which method to use it is important to understand the characteristics of the two systems. Each sperm nucleus is tightly associated to a centriole. In a very simple process, half spindles can assemble around sperm nuclei just by incubation of the sperm nuclei in extract. Over time these half spindles fuse to form bipolar spindles. If the extract is cycled through interphase the DNA replicates and each centrosome duplicates. The addition of fresh CSF extract sends the extract back into mitosis, chromosomes with their paired kinetochores condense, the duplicated centrosomes move apart, and a bipolar spindle assembles. Anaphase can be induced at this point by the addition of calcium to the system.
DNA beads are made from any kind of plasmid DNA that is long enough to induce chromatin assembly. They are unlikely to assemble kinetochores, as centromere sequences are not present. Spindle assembly around DNA beads is achieved by first assembling chromatin on the beads in an extract that is cycled through interphase. The beads are then retrieved and resuspended in fresh CSF extract. Using this method, spindles assemble in the absence of kinetochores and centrosomes. It is then ideal to study certain processes of spindle assembly that are independent of these structures.
A simplified method to study microtubule dynamics and focusing of microtubule minus ends is the assembly of asters in CSF extract. Asters can be assembled by the addition of human centrosomes purified from KE37 lymphoid cells (Bornens et al., 1987), DMSO, or taxol to the extract (Wittmann et al., 1998).
Solutions
- 10X calcium solution: 4 mM CaCl2, 100 mM KCl, 1 mM MgCl2. Store in aliquots at -20°C.
- 4.9M MgCl2 stock solution (Sigma)
- 10X MMR: See Section III,A.
- Hoechst dye solution: See Section III,B.
- Spindle fix: 48% glycerol, 11.1% formaldehyde, 5µg/ml Hoechst dye in 1X MMR. Always prepare fresh on day of use.
- BRB80: 80mM PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8. Prepare a 5X BRB80 solution. Dissolve components in sterile water and, while stirring, add KOH pellets until the PIPES dissolves. Adjust to pH 6.8 with KOH solution. Sterilize by filtration and store at 4°C.
- Dilution buffer: 1X BRB80 containing 30% glycerol and 1% Triton X-100. Store at room temperature. Better preservation of the spindles can be achieved by adding 0.25% glutaraldehyde to the dilution buffer. Use a full vial of glutaraldehyde and store in aliquots at -20°C.
- Aster dilution buffer: BRB80 containing 10% glycerol, 0.25% glutaraldehyde, 1 mM GTP, 0.1% Triton X- 100. Use a fresh vial of glutaraldehyde to prepare buffer and store in aliquots at -20°C.
- Cushion: 40% glycerol, BRB80. Store at 4°C.
- Aster cushion: 25% glycerol, BRB80. Store at 4°C.
- PBS: 137mM NaCl, 2.7mM KCl, 4.3mM Na2HPO4, 1.4 KH2PO4 mM, pH 7.4. Sterilize by autoclaving and store at room temperature.
- Immunofluorescence buffer (IF buffer): PBS containing 2% BSA and 0.1% Triton X-100. Add azide and store at 4°C.
- Mowiol: 10% Mowiol 4-88, 25% glycerol, and 0.1M Tris, pH 8.5. To prepare, mix 6g Mowiol with 6 g glycerol. Add 6 ml sterile water and stir for several hours at room temperature. Add 12ml 0.2M Tris, pH 8.5, and place the mixture in a heating plate at 50°C for 10min with continues stirring. After the Mowiol dissolves, centrifuge at 5000g for 15 min. Store in aliquots at -20°C.
Steps for Spindle Assembly around Sperm Nuclei (see Fig. 2)
- Before using an extract to assemble spindles, its quality should be tested by setting up a "half spindle" reaction. Combine 20 µl CSF extract, 0.2 µl rhodaminelabelled tubulin and 0.8µl sperm nuclei (about 75 nuclei/µl extract) in a 1.5-ml tube and mix. The extract can be mixed by gently moving the pipette in circles or by pipetting up and down, always avoiding making bubbles. Incubate the reaction mixture at 20°C and take a squash at 30-40min. To make a squash, using a cut-off tip, transfer 1 µl of reaction mixture to a microscope slide, carefully place 5 µl of spindle fix solution on top, and gently place a 18 × 18-mm coverslip on top. Analysis of the squash by fluorescence microscopy should reveal half spindles with condensed chromosomes and occasionally also spindles. There should be no free microtubule nucleation.
- If the extract is good, proceed with the spindle assembly protocol. For each reaction add on ice 0.2µl rhodamine-labelled tubulin and 0.8 µl sperm nuclei to 20 µl CSF extract in a 1.5-ml tube and mix. Incubate for 10 min at 20°C and then release extract into interphase by the addition of 2 g110X calcium solution. Mix gently.
- Incubate for 80min at 20°C. Check that the extract is in interphase by taking a squash as described in step 1. If the sample is to be saved, seal the coverslip to the slide with nail polish. At this stage, nuclei should appear large, round, and uniform, and microtubules should be long and abundant.
- At 90min postcalcium addition, add 20µl of fresh CSF extract containing 0.2 µl rhodamine-labelled tubulin to the reaction.
- Incubate further at 20°C. Take squashes (step 1) at different time points to assess the stage of spindle formation. During the incubation, prepare 15 ml Corex tubes with plastic adapters, a round 12-mm coverslip, and 5 ml of cushion.
- Forty-five to 60 min after mitosis reentry (step 4), quickly add 1ml dilution buffer and mix by gently inverting the tube a couple of times. Carefully layer the mixture over the cushion using a cut pipette tip. Centrifuge at 16°C for 12min at 12,000rpm in an HB-4 or HB-6 rotor. Aspirate supernatant and cushion before removing the coverslip. Fix coverslips in -20°C methanol for 5 minutes. Rehydrate samples by placing the coverslips in PBS. If dilution buffer contains glutaraldehyde, incubate the coverslips twice for 10min in 0.1% NaHB4 in PBS and wash with PBS.
- To perform immunofluorescence, place the coverslip face up on a surface covered with parafilm. Incubate the coverslips with primary antibodies diluted in IF buffer for 20-30 min. Wash the coverslips two times for 5 min with PBS and incubate with IF buffer containing the secondary antibody and 5µg/ml Hoechst for 20-30 min. Wash three times for 5 min with PBS and carefully place the coverslips upside down on a 3- to 4-µl drop of Mowiol on a microscope slide, avoiding air bubbles. Prior to observation, allow the Mowiol to set for 15min at 37°C or at room temperature for several hours.
- To visualize the early steps of anaphase, add 4 µl 10X calcium solution to the metaphase spindles before step 6. Take samples before calcium addition and every 5 min after calcium addition for 20-30min and process them as described earlier. It is not always possible to visualize separation of the sister chromatids and only high-quality extracts can be used successfully to study anaphase.
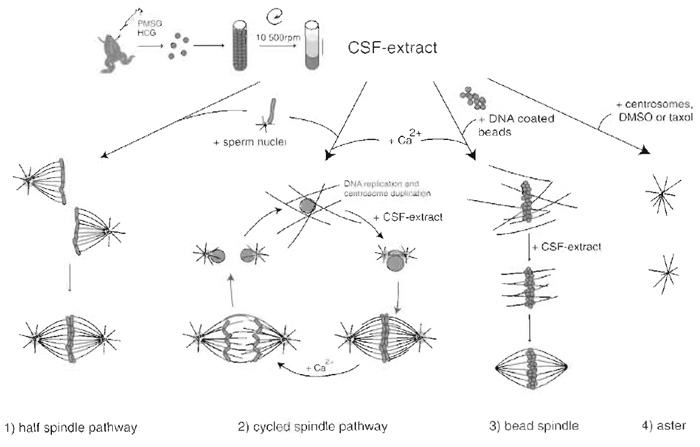 |
| FIGURE 2 Preparation of CSF Xenopus egg extract and structures that assemble after addition of different components. (1) Addition of sperm nuclei to the CSF extract leads to the formation of half spindles that can fuse to form bipolar spindles. (2) Addition of calcium together with sperm nuclei sends the system into interphase, allowing DNA replication and centrosome duplication to occur. Addition of fresh CSF extract sends the system back to mitosis and bipolar spindles form. Calcium addition at this point induces chromosome segregation and entry into interphase. (3) Bead spindle: addition of DNA-coated beads and calcium to an extract leads to the formation of chromatin and nuclei around beads. Addition of CSF extract followed by incubation in fresh CSF extract allows the formation of bipolar spindle. (4) Asters: Addition of purified centrosomes or DMSO or taxol leads to the formation of microtubule asters with microtubule minus ends focused at the centrosome or at the center of the aster. |
Steps for Spindle Assembly around Chromatin Beads (see Fig. 2)
- Transfer 3 µl of DNA beads (about 0.5 µg DNA) to a 0.5-ml Eppendorf tube and place on a magnet. Remove supernatant, and wash beads by resuspending them in 20 µl of extract.
- Retrieve beads and resuspend in 100µl CSF extract. Transfer to a 1.5-ml Eppendorf tube and incubate at 20°C.
- After 10min, release the CSF extract into interphase by adding 10µl of calcium solution. Incubate for 2h at 20°C.
- Return the extract containing the beads to mitosis by adding 50µl of fresh CSF extract. Incubate for 30 more min at 20°C. The procedure can be continued or stopped at this point by freezing the beads in the extract using liquid ethane and storing them in liquid nitrogen.
- Incubate the bead mixture on ice for several minutes. Retrieve the beads on ice over 10-15 minutes. Due to the viscosity of the extract, bead retrieval is slow. Pipette the mixture every several minutes, keeping the tube on the magnet.
- Remove the supernatant and resuspend the beads in 150µl of fresh CSF extract containing 1.5µl rhodamine-labelled tubulin.
- Incubate at 20°C. Monitor the spindle assembly by taking 1-µl samples and squashing with fixative as described earlier. Spindle assembly requires between 30 and 90 min, depending on the extract. For immunofluorescence studies, spin the bead spindles through a cushion onto a coverslip and perform immunofluorescence as described previously for spindle assembled around sperm nuclei.
Steps for Aster Formation (see Fig. 2)
- For each reaction, add on ice 0.2µl rhodaminelabelled tubulin to 20µl CSF extract in a 1.5-ml tube. To assemble asters, add either purified human centrosomes (Bornens et al., 1987) or 5% DMSO or 1 µM taxol.
- Incubate the reaction for 30-60min in a 20°C. water bath. During this incubation, prepare as before 15-ml Corex tubes with plastic adapters, round 12-mm coverslip, and 5 ml aster cushion.
- At the end of the incubation time, dilute the reactions with aster dilution buffer and carefully layer them on top of the cushion using a pipette with a cut tip and subsequently centrifuge onto the coverslips in a HB4 or HB6 rotor at 12,000rpm for 12min at 16°C.
- As before, remove the cushion with a vacuum pump and postfix the coverslips in -20°C methanol for 5min. To quench the glutaraldehyde, incubate twice for 10min in 0.1% NaHB4 in PBS and process for immunofluorescence.
D. Functional Studies of Proteins Involved in Spindle Assembly
The Xenopus egg extract system is a powerful tool to assess the involvement of individual proteins in the process of spindle assembly. Different methods have been developed to address specific questions.
Localization studies can be performed by classical immunofluorescence methods or by direct visualization of GFP-tagged proteins added to the reaction mixture or by a combination of both by adding GSTtagged proteins followed by immunofluorescence with anti-GST antibodies.
Functional studies can be performed by adding dominant-negative constructs to the reaction mixture or by depleting the protein or antibodies under study from the extracts before use (Antonio et al., 200··; Boleti et al., 1996). Depletion experiments should be complemented by "add back" or rescue experiments in which a recombinant protein is added to the depleted extracts (Fig. 3). In all tree cases, a careful quantification of the structures found in control or treated samples has to be performed.
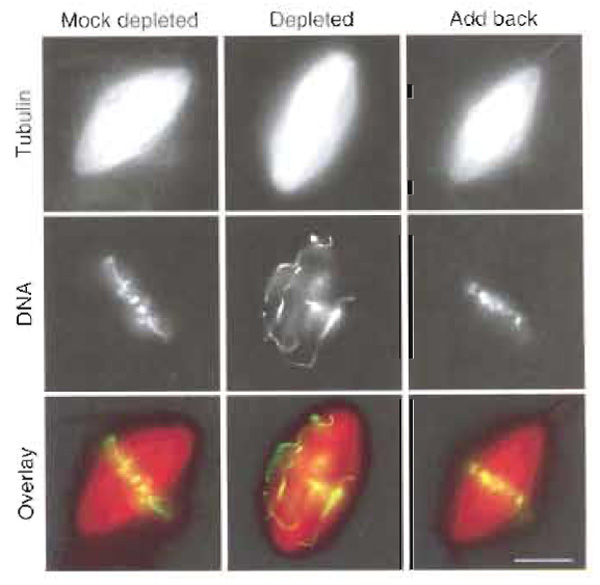 |
| FIGURE 3 Spindles assembled in CSF Xenopus egg extract following the cycled spindle pathway. The extract was mock depleted, Xkid depleted, or Xkid depleted supplemented with recombinant Xkid protein (add back). When Xkid was depleted, mitotic chromosomes failed to align at the metaphase plate. Alignment of chromosomes was rescued by the addition of recombinant protein to the extract. Scale bar: 10 µm. |
Solutions
- CSF-XB with PI: See Section III,A.
- PBS-TX: PBS containing 0.1% Triton X-100. Store at 4°C.
Steps for Immunodepletion
- Usually around 10µg of specific antibody is needed to deplete 150 µl of extract. The amount of antibody has to be adjusted for each case depending on the antibody itself and on the abundance of the protein in the extract. Prepare a control mock-depleted extract using the same amount of unspecific purified IgG.
- Transfer 40 µl of Dynal beads coupled to protein A to a 1.5-ml tube and retrieve them using a magnet. Wash them twice by resuspending the beads in 500µl PBS-TX and retrieving them with the magnet.
- Incubate the beads with 10 µg of antibody diluted in 200 µl of PBS-TX for 1h on a rotating wheel at 4°C.
- Wash twice as just described with PBS-TX and once with CSF-XB containing PI. Remove as much wash buffer as possible and carefully resuspend the beads in 150µl of extract by pipetting up and down. Keep the extract (containing the beads) on ice for 90min with occasional mixing by pipetting up and down.
- Retrieve the beads by placing the tube on the magnet for 5 min on ice. Collect the extract into a new tube and keep it on ice. To remove the beads more efficiently, repeat this procedure once more. Check the efficiency of depletion by Western blot analysis of mock and protein-depleted extracts. To visualize the immunoprecipitated protein, wash the beads once with CSF-XB containing PI and three times with PBSTX. Resuspend the beads in Laemmli sample buffer and incubate at room temperature for 5 min. Retrieve the beads on a magnet and boil the supernatant before loading on SDS-PAGE.
- An important control to include in depletion experiments is the add back of the purified recombinant protein to the depleted extract in a range of concentrations close to that of the endogenous protein. In case the protein is degraded during interphase, the recombinant protein should be added only at mitosis reentry.
- The depleted, the control mock depleted, and add-back extracts can now be used in parallel to assemble asters and spindles following the methods described in Section III,C.
- Compare the spindles or asters formed in each reaction either by taking samples and squashing them in a slide with fixative solution or by centrifuging them onto coverslips and performing immunofluorescence.
Steps for Specific Antibodies or Dominant-Negative Protein Addition
- In the case of addition of specific antibodies, prepare a concentrated solution (at least 2mg/ml) of an affinity-purified polyclonal or monoclonal antibody in PBS or CSF-XB. It is a good idea to try different concentrations of antibody (ranging from 200 to 400µg/ ml). For dominant-negative addition, prepare constructs expressing different fragments of the protein under study fused to GFP or GST. Purify the fusion proteins and dialyze them extensively against CSF-XB.
- Add the antibodies or dominant-negative protein to the extract in a volume corresponding to one-tenth of the total volume and follow the protocol for spindle assembly or aster formation as described in Section III,C. The antibodies or dominant negative can be added at the beginning of the spindle assembly procedure or at mitosis reentry.
- When adding antibodies, two control samples should be run in parallel: one containing a similar amount of a control antibody and another containing the same volume of buffer. In the case of dominantnegative addition, include also the same volume of buffer as control and a sample containing GST or GFP protein.
- Samples taken at different time points are fixed and analyzed as before. To examine the localization of the antibody, process the samples by immunofluorescence using a fluorochrome-conjugated secondary antibody. Localization of the dominant negative can be observed directly if the tag is GFP or by using an anti- GST antibody.
IV. PITFALLS
The biggest pitfall in studying spindle assembly with this system is the problem of reproducibility in Xenopus egg extracts. To obtain good eggs, the frog colony must be healthy and well cared for. This requires a substantial commitment on the part of the laboratory. Even with healthy frogs, there is seasonal variation in the quality of eggs, with summer being the off season. Furthermore, even experienced extract makers do not manage to prepare functional extracts every time, which can be frustrating. Therefore, experiments must be repeated several times to ensure a valid interpretation.
References
Antonio, C., Ferby, I., Wilhelm, H., Jones, M., Karsenti, E., Nebreda, A. R., and Vernos, I. (2000). Xkid, a chromokinesin required for chromosome alignment on the metaphase plate. Cell 102(4), 425-435.
Boleti, H., Karsenti, E., and Vernos, I. (1996). Xklp2, a novel Xenopus centrosomal kinesin-like protein required for centrosome separation during mitosis. Cell 84, 49-59.
Bornens, M., Paintrand, M., Berges, J., Marty, M. C., and Karsenti, E. (1987). Structural and chemical characterization of isolated centrosomes. Cell Motil. CytoskeI. 8(3), 238-249.
Desai, A., Maddox, P. S., Mitchison, T. J., and Salmon, E. D. (1998). Anaphase A chromosome movement and poleward spindle microtubule flux occur at similar rates in Xenopus extract spindles. J. Cell Biol. 141(3), 703-713.
Desai, A., Murray, A., Mitchison, T. J., and Walczak, C. E. (1999). The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro. Methods Ceil Biol. 61, 385-412.
Evans, L., Mitchison, T. J., and Kirschner, M. W. (1985). Influence of the centrosome on the structure of nucleated microtubules. J. Cell Biol. 100, 1185-1191.
Gurdon, J. B. (1976). Injected nuclei in frog oocytes: Fate, enlargement, and chromatin dispersal. J. Embryol. Exp. Morphol. 36, 523-540.
Heald, R., Tournebize, R., Blank, T., Sandaltzopoulos, R., Becker, P., Hyman, A., and Karsenti, E. (1996). Self organization of microtubules into bipolar spindles around artificial chromosomes in Xenopus egg extracts. Nature 382, 420-425.
Hyman, A., Drechsel, D., Kellogg, D., Salser, S., Sawin, K., Steffen, P., Wordeman, L., and Mitchison, T. (1991). Preparation of modified tubulins. Methods Enzymol. 196, 478-485.
Kalab, P., Weis, K., and Heald, R. (2002). Visualization of a Ran-GTP gradient in interphase and mitotic Xenopus egg extracts. Science 295(5564), 2452-2456.
Lohka, M., and Maller, J. (1985). Induction of nuclear envelope breakdown, chromosome condensation, and spindle formation in cell-free extracts. J. Cell Biol. 101, 518-523.
Murray, A. (1991). Cell cycle extracts. In "Methods in Cell Biology" (B. K. Kay, H. B. Peng, eds.), vol. 36, pp. 581-605. Academic Press, San Diego.
Murray, A. W., and Kirschner, M. W. (1989). Cyclin synthesis drives the early embryonic cell cycle. Nature 339, 275-280.
two different pathways in vitro. J. Cell Biol. 112, 925-940.
Shamu, C. E., and Murray, A. W. (1992). Sister chromatid separation in frog egg extracts requires DNA topoisomerase II activity during anaphase. J. Cell Biol. 117, 921-934.
Wittmann, T., Boleti, H., Antony, C., Karsenti, E., and Vemos, I. (1998). Localization of the kinesin-like protein Xklp2 to spindle poles requires a leucine zipper, a microtubule-associate'd protein, and dynein. J. Cell Biol. 143(3), 673-685.
Support our developers




