Induction of Differentiation and Cellular Manipulation of Human Myeloid HL-60 Leukemia Cells
I. INTRODUCTIONThe human myeloid HL-60 leukemia cell line has enjoyed widespread use in studies of the molecular mechanisms involved in the control of cell growth, differentiation, and apoptosis. Following stimulation with specific agents, these cells acquire a granulocytic (Collins et al., 1980), monocytic, or macrophage-like phenotype (Murao et al., 1983). The maturation of these precursor cells into their mature progeny occurs at a high percentage and with some degree of predictable temporal uniformity. The acquisition of mature phenotypes can be demonstrated by a variety of differentiation markers. These include enzymatic markers such as an increase in the activities of nonspecific esterase or lysozyme; morphological changes such as the appearance of segmented nuclei, which are characteristic of a granulocyte-like phenotype (Collins et al., 1980); or other markers such as cell attachment and spreading on culture dishes or increased phagocytosis, which are indicators of a macrophage phenotype (Tonetti et al., 1994). Additionally, there are a variety of well-characterized immunological markers, some of which are lineage and/or stage specific. The use of these markers and forceful expression of specific cDNAs or inhibition of a particular gene's expression by pertinent antisense oligonucleotides has allowed for the identification of the temporal order of many components involved in the signal transduction processes that lead to HL-60 cell differentiation (Semizarov et al., 1998; Laouar et al., 1999; Xie et al., 1998ab).
II. MATERIALS AND INSTRUMENTATION
HL-60 cells (Cat. No. CCL-240) are available from the American Type Culture Collection (ATCC). Culture medium RPMI 1640 (Cat. No. 11875-093), penicillin-streptomycin-glutamine (Cat. No. 10378- 016), geneticin (Cat. No. 10131-035), and trypsin- EDTA (Cat. No. 15400-054) are from Invitrogen Life Technologies. Phorbol 12-myristate 13-acetate (PMA; Cat. No. P-1680) is from L.C. Laboratories, Inc. All-trans retinoic acid (ATRA; Cat. No. R-2625), L-0Mysophosphatidylcholine (Cat. No. L-4129), paraformaldehyde (Cat. No. P-1648), bovine serum albumin (BSA) fraction V (Cat. No. A-7906), anti-cdllb (Cat. No. C-0051), transferrin (Cat. No. T-2036), and insulin (Cat. No. 1-6634) are available from Sigma Chemical Co. 1α,25-Dihydroxy-vitamin D3 [1,25-(OH)2vitD3; Cat. No. 679101] and hygromycin B (Cat. No. 400050) are from CalBiochem. PolyMount solution (Cat. No. 16866), 1.72-µm fluoresbrite beads (Cat. No. 17687), and hydroethidine (Cat. No. 17084) are from Polysciences Inc. 4',6-Diamidine-2'-phenylindole dihydrochloride (DAPI; Cat. No. 236276) is available from Boehringer Manneheim Biochemicals. Lab-Tek chamber slides (eight-well, Cat. No. 178599) are from Nunc, Inc. Fetal bovine serum (FBS; Cat. No. SH30071) is from HyClone. Dimethyl sulfoxide (DMSO) molecular biology grade (Cat. No. BP231-100) is from Fisher. Antibodies against CD14 (Cat. No. 30541A) and HLA-DR (Cat. No. 555562) are available from BD Pharmingen. Antibody against human glyceraldehde-3-phosphate dehydrogenase (GAPDH, Cat. No. CR1093SP) is from Cortex Biochem. Antibody against actin (Cat. No. sc-1615) is from Santa Cruz Biotech. Vector pRL-null (Cat. No. E227A) is from Promega, and pIRES2-EGFP (Cat. No. 6029-1) is from BD Clontech.
III. PROCEDURES
A. Cell Growth and Differentiation Induction
Materials
- Culture medium: Supplement RPMI 1640 culture medium with penicillin-streptomycin (100µg/ml), L-glutamine (200mM), and 10% FBS. If cells are to be used for viable immunostaining, inactivate serum complement by heat inactivation (65°C for 15 min followed by slow cooling to room temperature). Culture medium can be stored at 4°C for several months.
- Differentiation inducer stock solutions: Dissolve PMA, 1,25-(OH)2vitD3, and ATRA in DMSO at a concentration of 1 mg/ml. Store small aliquots in sterile microcentrifuge tubes at-70°C and avoid repeated freezing/thawing.
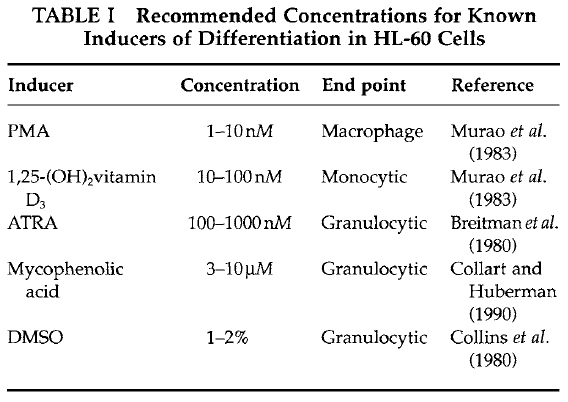
- Differentiation inducer working solutions: The most effective concentration for the induction of differentiation depends on the potency of individual lots of the inducer and the endogenous sensitivity of the HL-60 cells being used. Examples of appropriate concentrations for some common inducers of HL-60 differentiation are shown in Table I. For new chemical inducers, a series of concentrations of the inducer should be tested, and conditions that impart an inhibition of cell multiplication and the appearance of one or more myelomonocytic differentiation markers should be tested further. For many differentiation inducers, cells will exhibit maturation markers at inducer concentrations that inhibit about 50% or greater of cell multiplication rates.
- Cell culture: The HL-60 cell line has a population doubling time of 20-24 h. Cell density should be maintained between 2 × 104 and 1 × 106 cells/ml with replacement of medium at high densities by pelleting of the cells by centrifugation if medium becomes acidified. Cultures should be maintained in either petri dishes or tissue culture plates at 37°C in an 8% CO2, 95% water-humidified atmosphere.
 |
Steps
- Collect the cells and obtain cell density by hemocytometer chamber counting. Pellet the cells by gentle (250g) centrifugation. Resuspend in fresh growth medium and plate the cells at 2-10 × 104 cells/ml in culture dishes. Allow the cells to recover for several hours prior to addition of the inducing agent.
- Dilute the differentiation-inducing agent in a minimal volume of culture medium and add to the experimental plates. Be sure to include a control culture to which the solution vehicle alone is added.
- Culture cells at 37°C in an 8% CO2 humidified atmosphere.
- The appearance of differentiation markers occurs
as early as 12h and up to 6 days after treatment,
depending on the concentration of the inducing agent
and the marker to be assessed. To evaluate the phenotypic
changes associated with cell differentiation, a
series of morphological, biochemical, and immunological
assays are performed (see Table II). We recommend that two to three different assays be used to
determine whether the cells acquire a granulocytic,
monocytic, or macrophage-like phenotype. Many of the biochemical or histochemical markers can be assessed using commercially available kits.
 |
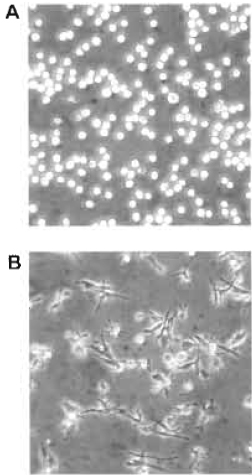 |
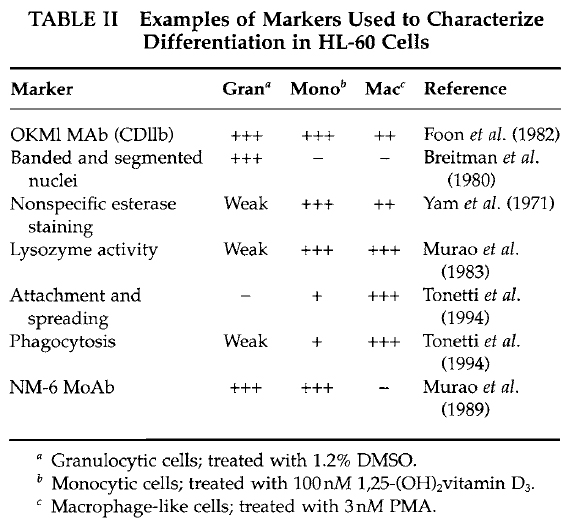
| FIGURE 1 Bright-field microscopy images representing an example of HL-60 cells either untreated in suspension (A) or attached and spread following a 3-day treatment with 10nM PMA (B). |
Material
Phosphate-buffered saline (PBS): To make 1 liter of stock solution (10×), dissolve 80.0 g of NaCl, 2.0 g of KCl, 2.0 g of KH2PO4, and 11.4 g of Na2HPO4 in highquality (Milli-Q) water and sterilize by autoclaving. Dilute to l× in water.
Steps
- Treat HL-60 cells cultured in bacteriological petri dishes (see Section IV) with PMA and incubate at 37°C in an 8% CO2, humidified atmosphere for 2 days.
- Collect unattached cells by gentle pipetting and wash petri dishes twice with PBS, combining the washes with the initially collected cells.
- Recover the remaining attached cells by treatment for 10rain with PBS supplemented with 0.05% trypsin-EDTA followed by forceful pipetting and/or use of a rubber policeman.
- Count the number of attached and unattached cells using a hemocytometer chamber and calculate the percentage of attached cells as a function of the total recovered cell number.
- In parallel, determine the number of spread cells by counting flattened cells (Fig. 1) in several microscopic fields of view. Count at least 200 cells/data point; the percentage cell spreading = (number of spread cells/total number of adherent cells) × 100.
C. Phagocytosis Assay
Materials
- Fluorescent beads: Sterilize and opsonize fluoresbrite beads by first adding three drops of beads to 10ml of 70% ethanol. Mix and agitate at room temperature for 20min. Recover the beads by centrifugation (400g) and wash twice with PBS. After the second wash, add 5 ml of RPMI 1640 supplemented with 1 ml FBS (not heat inactivated). Mix and incubate overnight at 37°C with slow agitation. Pellet the beads at 1000g and resuspend in 1 ml of culture medium at a concentration of 4 × 108 beads/ml.
- Working solutions: Prepare the following solutions using PBS as solvent: 4% paraformaldehyde, 10 µg/ml L-α-lysophosphatidylcholine, 0.1 µg/ml DAPI, and 10 µg/ml hydroethidine.
Steps
- Plate 1.2 × 104 cells in 400µl of growth medium into each well of an eight-well Lab-Tek chamber slide. Treat the cells with PMA or other inducer as described previously.
- After 30h, add 100µl of the bead suspension to each well.
- Incubate for an additional 18h, gently remove the growth medium, and air dry the cells for 30 s using a heated slide dryer.
- Fix the cells by adding 100µl of 4% paraformaldehyde to each well for 20min and then washing each well several times with PBS.
- Add 100 µl of L-α-lysophosphatidylcholine solution to each well for 20min to permeabilize the cells, followed by several washes with PBS.
- Cell nuclei are stained by incubation with DAPI for 5 min. Remove excess stain by washing with PBS and then counterstain the cytoplasm by incubating with hydroethidine for 10 min.
- After three washes in PBS, remove the well partitions and mount the slide with Poly-Mount or other mounting medium and a coverslip.
- Examine the cells using a fluorescent microscope equipped with a DAPI/UV-range filter set. DAPIstained nuclei should appear blue, the stained cytoplasm red, and the fluoresbrite beads as small blue dots. A cell is considered positive for phagocytosis activity if it has engulfed more than 20 beads. Count >200 cells per point.
D. Immunofluorescence
Materials
- PBSA: dissolve BSA (Cohn fraction V) to 1% in PBS.
- Primary antibodies: Numerous monoclonal and polyclonal antibodies are available to characterize the phenotype of mature myeloid cells. These include but are not limited to the Mac-1/antiCDllb, CD14, or class II HLA-DR antigens. A more categorical listing is available online (http://www.bdbiosciences.com/pdfs/ other/01-81024-3.pdf). These are available from many commercial sources (e.g., Immunotech, Coulter Immunology, Santa Cruz, Sigma, Pharmingen) and should be prepared and used according to the manufacturer's recommendations. For a new antibody, it is often prudent to perform a dilution series with both positive and negative controls to select an antibody concentration that provides a positive signal with a low background. For semiquantitative ratio imaging, useful standard antibodies are against GAPDH (sheep polyclonal, Cortex Biochem.) or actin (goat polyclonal, Santa Cruz). All antibody stock solutions are created by dilution in PBSA.
- Secondary antibodies: There are numerous sources of labeled secondary antibodies. If one is to perform multiantigen imaging, it is critical that the secondary antibodies have been cross-absorbed against the other host organisms antibody repertoire. Such crossabsorbed antibodies are available tagged to a variety of fluorophores from both Jackson Immunoresearch and Molecular Probes. Stock solutions of these secondary antibodies are prepared at 1 mg/ml in 50% glycerol/50% PBS. Store aliquots of the stock solution at-70°C. Prepare a working solution of the secondary antibody by diluting from 1:50 to 1:500 in PBSA using the furthest dilution that still provides significant signal.
- Microscopy: A microscope equipped with multiple filter sets and/or cubes is required to visualize the fluorescence signal from multiple fluorophores. If one is to perform semiquantitative ratio imaging using multiple fluorescent signals, it is useful to have either an automated cube turret or automated excitation and emission filter wheels and a multipass dichroic cube. Both this hardware and SlideBook software for processing such images are available from Intelligent Imaging Innovations (Denver, CO).
- Flow cytometery: For the assessment of marker induction in granulocytic differentiation, flow cytometry has many advantages, particularly regarding the stastical significance attainable by scanning larger cell populations. However, in the case of monocytic and macrophage differentiation, significant clumping of cells is unavoidable and may lead to artifactual results. We therefore recommend fluorescence microscopy for such experiments and the examination of at least 200 cells per data point.
Steps
Reactions are performed on ice using either microcentrifuge tubes or, to facilitate the handling of numerous
samples simultaneously, V-bottom 96-well plates.
- Recover the cells by centrifugation and resuspend in PBS at 2 × 106/ml; add 200µl of the cells for each collection of antibodies to a well of the microtiter plate.
- Centrifuge the plate (400g), remove the supernatant gently by inversion, and fix the cells by incubation with 100µl of 4% paraformaldehyde for 15min. Wash twice with PBS.
- If the antigen to be visualized is not on the outside of the cell membrane, permeabilize the cells by the addition of either L-α-lysophosphatidylcholine solution or 0.1% Triton X-100 in PBS for 5min. Wash the cells three times with PBS.
- Resuspend the cells in 100 µl of PBSA and allow for blocking of nonspecific sites by incubation for 15min. Recover cells by centrifugation and mix the cells with 100µl of diluted primary antibody or mixture of antibodies (or a dilution series may be used) and incubate for 45 min at room temperature.
- After washing the cells twice with PBS, add 100µl of diluted secondary antibodies and continue incubating for 30min.
- Wash the cells three times with PBS and then mount onto microscope slides using a small volume of a mounting solution. Overlay with a cover glass and seal with nail polish. Examine by fluorescence microscopy.
E. Transfection and Establishment of Stable Cell Lines
Materials
- Most chemical approaches (e.g., charged lipids) for the transfection of HL-60 cells produce very poor results. Better transfection is achieved using electroporation, particularly with a square-wave device, and to a lesser degree by an exponential-decay device. We use a BTX T820 square-wave electroporator and achieve maximally 10-20% transfection rates.
- For analysis of promoter activities, either CAT assays or luciferase activities can be monitored in HL-60 cells. However, standard firefly luciferase is unstable or degrades rapidly and the Renilla luciferase should be employed instead (pRL vectors, Promega).
- Due to the low transfection efficiency of HL-60 cells, transient transfection assays should include a readily visualizable marker of transfection such as cotransfection with an EGFP plasmid, or preferably using a vector that expresses both your gene of interest and EGFP from the same plasmid (pIRES2-EGFP, Clontech).
Steps
- If transient assays are to be performed, use 15 btg of supercoiled plasmid per transfection. If stable transfectants are to be isolated, linearize each plasmid with a restriction endonuclease that cleaves the vector but leaves your gene of interest, the selectable marker, and any regulatory elements intact. Solve either preparation in a small volume of PBS.
- Collect HL-60 cells by centrifugation and resuspend at 1.0 × 106 in growth medium. Combine 400µl of cells with DNA in a 0.4-cm-gap electroporation cuvette. Allow to stand for 5 min at room temperature.
- Mix the cuvette by gentle tapping and insert into a square-wave electroporation device. Electroporate with three pulses of 1500 V each and 90µs duration.
- Allow the cells to recover for 5 min and then plate into 10ml of growth medium and incubate at 37°C in 8% CO2 in a humidified atmosphere.
- For transient assays, recover the cells the next day and perform CAT or luciferase assays by standard protocols. For monitoring of fluorescence from EGFP expression, prepare the cells as in steps 1 and 2 of the immunofluorescence protocol, mount the cells, and examine for EGFP expression using a FITC filter set.
- For stable transfectant recovery, wait 24h and then add appropriate selective agent. Geneticin and hygromycin B are effective at 500 and 150µg/ml, respectively, in HL-60 cells.
- Allow for selection of stable transfectants by growth for 7-10 days. Cells can either be maintained at this stage as a pooled group of transfectants or individual clones can be isolated by single-cell dilution and expansion in 96-well plates.
- Successful transfection in pools or individual clones should be monitored either by immunofluorescence or by Western blotting using either an antibody against the protein or an epitope tag if present.
F. Inhibition of Gene Expression Using Synthetic Oligonucleotides
Materials
- Synthetic oligonucleotides should be synthesized as 15-mers using standard base chemistry. The exact concentration at which an oligonucleotide effectively disrupts translation or targets a given mRNA for RNase-mediated decay is a function of intracel-lular concentration, expression level of the target, and ability of the oligonucleotide to hybridize, i.e., secondary structure limitations. Therefore, several oligonucleotides and a range of oligonucleotide concentrations with a suitable scrambled control oligonucleotide need to be empirically tested. Initial experiments with siRNA indicate that effective gene knockdown by this approach can be performed transiently, but long-term expression of dsRNA may induce an interferon response (Bridge et al., 2003) in HL-60 cells.
- Prepare solutions of 5mg/ml transferrin and 5mg/ml insulin in unsupplemented RPMI 1640 medium. Prepare serum-free growth medium by supplementing RPMI 1640 with standard concentrations of penicillin-streptomycin (100µg/ml) and L-glutamine (200 mM).
Steps
- Collect HL-60 cells by centrifugation and wash three times in serum-free growth medium. Count the cells by hemocytometer chamber counting and plate the cells at 1 × 105 cells/ml in serum-free growth medium.
- Supplement the cells with 5µg/ml transferrin and 5 µ/ml insulin. Add a series of dilutions of your test and control oligonucleotides spanning a concentration range from 100 nM to 100 µM.
- Allow uptake of the oligonucleotide to occur over a period of 6h incubation at 37°C in an 8% CO2 in a humidified atmosphere.
- Restore FBS levels to 10% and allow overnight growth.
- Monitor toxicity to the cells by trypan blue dye exclusion assays. Determine the effect of your test and control oligonucleotides on the expression of your gene of interest by either immunofluorescence or reverse transcriptase polymerase chain reaction (rtPCR) approaches.
- Subsequent effects on differentiation induction and specific marker appearance can then be determined by following steps 1-4 and then adding the specific chemical inducer.
IV. COMMENTS AND PITFALLS
- When thawing materials (antibodies, antigen, or samples), mix thoroughly before dilution or processing. Avoid multiple freeze-thaw cycles by aliquoting small volumes of reagents into multiple microcentrifuge tubes for storage.
- When using all-trans retinoic acid, avoid exposure to light.
- Fluorescent-activated cell sorting quantitation is not recommended when the differentiation inducer causes cell clumping, as is the case with PMA and related chemicals.
- For some inducers that cause attachment, trypsin-EDTA treatment may not remove all cells from the surface of tissue culture dishes. Therefore, bacteriological grade petri dishes are utilized for procedures that require removal of the attached cells.
- Use multiple differentiation markers when testing the effect of a new inducer.
- In preparing cells for immunological analysis, use heat-inactivated serum to eliminate cell killing due to active complement.
Acknowledgments
This work was supported by the U. S. Department of Energy, Office of Health and Environmental Research, under Contract W-31-109-ENG-38, and the National Institutes of Health under Grant CA80826.
References
Breitman, T. R., Selonick, S. E., and Collins, S. I. (1980). Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 77, 2936-2940.
Bridge, A. J., Pebernard, S., Ducraux, A., Nicoulaz, A. L., and Iggo, R. (2003). Induction of an interferon response by RNAi vectors in mammalian cells. Nature Genet. 34, 263-264.
Collart, F., and Huberman, E. (1990). Expression of IMP dehydrogenase in differentiating HL-60 cells. Blood 75, 570-576.
Collins, S. I., Bonder, A., Ting, R., and Gallo, R. C. (1980). Induction of morphological and functional differentiation of human promyelocytic leukemia cells (HL-60) by compounds which induce differentiation of murine leukemia cells. Int. J. Cancer 25, 213-218.
Foon, K. A., Schroff, R. W., and Gale, R. P. (1982). Surface markers on leukemia and,lymphoma: Recent advances. Blood 60, 1-19.
Laouar, A., Collart, E R., Chubb, C. B., Xie, B., and Huberman, E. (1999). Interaction between alpha 5 beta 1 integrin and secreted fibronectin is involved in macrophage differentiation of human HL-60 myeloid leukemia cells. J. Immunol. 162, 407-414.
Murao, S., Collart, E R., and Huberman, E. (1989). A protein containing the cystic fibrosis antigens is an inhibitor of protein kinases. J. Biol. Chem. 264, 8356-8360.
Murao, S., Gemmell, M. A., Callallam, M. E, Anderson, N. L., and Huberman, E. (1983). Control of macrophage cell differentiation in human promyelocytic HL-60 leukemia cells by 1,25-dihydroxyvitamin D3 and phorbol-12-myristate-13-acetate. Cancer Res. 43, 4989-4996.
Semizarov, D., Glesne, D., Laouar, A., Schiebel, K., and Huberman, E. (1998). A lineage-specific protein kinase crucial for myeloid maturation. Proc. Natl. Acad. Sci. USA. 95, 15412-15417.
Tonetti, D. A., Henning-Chubb, C., Yamanishi, D. T., and Huberman, E. (1994). Protein kinase C-β is required for macrophage differentiation of human HL-60 leukemia cells. J. Biol. Chem. 269, 23230-23235.
Xie, B., Laouar, A., and Huberman, E. (1998a). Autocrine regulation of macrophage differentiation and 92-kDa gelatinase production by tumor necrosis factor-alpha via alpha5 betal integrin in HL- 60 cells. J. Biol. Chem. 273, 11583-11588.
Xie, B., Laouar, A., and Huberman, E. (1998b). Fibronectin-mediated cell adhesion is required for induction of 92-kDa type IV collagenase/ gelatinase (MMP-9) gene expression during macrophage differentiation: The signaling role of protein kinase C-beta. J. Biol. Chem. 273, 11576-11582.
Yam, L., Li, C. Y., and Crosby, W. H. (1971). Cytochemical identification of monocytes and granulocytes. Am. J. Clin. Pathol. 55, 283-290.
Support our developers




