Isolation of Mitochondria from Mammalian Tissues and Cultured Cells
I. INTRODUCTIONIsolation of mitochondria can be a necessary procedure for many purposes: (1) as a primary step for further purification of mitochondrial subcomponents; (2) to perform metabolic assays, with respiration activity analysis the most common; and (3) to perform molecular analyses on the biogenetic activity of the organelle. The mitochondrial preparations obtained using the method described herein are perfectly suitable for biogenetical studies (mitochondrial DNA, RNA, protein synthesis, and as source for respiratory complex analysis by blue-native gel electrophoresis), as well as in a variety of different assays where isolated mitochondria are required, such as assessment of respiratory enzyme activities, protein import, aminoacylation, and in organello footprinting. The method consists of three basic steps: (1) cell rupture, (2) differential centrifugation: first at low speed to pellet mainly nuclei and unbroken cells and then at high speed to pellet mitochondria, and (3) washing of the mitochondrial pellet in order to reduce the presence of other subcellular contaminants. Mitochondria from different sources are obtained using basically the same methodology, although some modifications must be introduced depending on the tissue type or if mitochondria are to be isolated from cultured cells. When the purity of samples is considered, there is an elimination of a good part of the contaminants when mitochondrial preparations obtained by this procedure are compared with crude mitochondrial fractions. Because this purification protocol does not make use of gradient preparation or ultracentrifugation, further purification of the organelles is recommended when mitochondria are prepared for isolation of organelle subcomponents. In summary, the method described here produces reasonably pure mitochondria in a fairly short time and with a low cost. In addition, maintenance of integrity and functionality of the organelles are guaranteed.
II. MATERIALS AND INSTRUMENTATION
Sucrose (ACS for analysis) is from Carlo Erba Reagenti (Cat. No. 477183). Sodium chloride (ACS analytical reagent, Cat. No. 727 810) is from Prolabo. Potassium cyanide (KCN; BioChemika MicroSelect; Cat No. 60178) is from Fluka. Ethylenediaminetetraacetic acid disodium salt 2-hydrate (EDTA-Na2; for analysis-ACS; Cat. No. 131669), potassium chloride (for analysis-ACS-ISO; Cat. No. 131494), magnesium chloride 6-hydrate (for analysis-ACS-ISO; Cat. No. 131396), sulphuric acid (96%; Cat. No. 251058), trichloroacetic acid [solution 20% (w/v); Cat. No. 252373], acetic acid glacial (chemically pure; Cat. No. 211008), sodium hydrogen sulphite [solution 40% (w/v); Cat. No. 211642], sodium sulphite (anhydrous purissimum; Cat. No. 141717); dipotassium hydrogen phosphate (anhydrous for analysis; Cat. No. 121512), potassium dihydrogen phosphate (for analysis, Cat. No. 121509) and orthophosphoric acid (85% for analysis-ACS-ISO; Cat. No. 131032) are from Panreac. D-Mannitol (ACS reagent; Cat. No. M-9647), ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA; approximately 97%; Cat. No. E-4378), Lglutamic acid monosodium salt [glutamate; minimum 99% (TLC); Cat. No. G-1626], L-(-)-malic acid sodium salt [malate; 95-100% (enzymatic); Cat. No. M-1125], succinic acid disodium salt hexahydrate (succinate; minimum 99%; Cat. No. S-2378), oxalacetic acid (approximately 98%; Cat. No. O-4126), acetyl coenzyme A trilithium salt (acetyl-CoA; approximately 95%, Cat. No. A-2181), 5-5'-ditio-bis(2-nitrobenzoic acid) or DTNB (Ellman's reagent; Cat. No. D-8130), glycerol 2-phosphate disodium salt hydrate (β- glycerophosphate; ≤ 0.1% α-isomer; Cat. No. G-6251), ammonium molybdate tetrahydrate (81-83% as MoO3 ACS reagent; Cat. No. A-7302), β-D-glucose 6- phosphate sodium salt (crystalline, Sigma Grade; G- 7879), imidazole [minimum 99% (titration), crystalline; Cat. No. 1-0250], hydrogen peroxide (29-32% as H2O2 ACS reagent; Cat. No. H-0904), bovine serum albumin [BSA; fraction V; minimum 96% (electrophoresis); Cat. No. A-4503], and subtilisin (subtilisin A from Bacillus sp.; lyophilized powder, type VIII, 7-15 units/mg solid; Cat. No. P-5380) are from Sigma. Aldrich supplied the titanium (IV) oxysulfate [titanyl sulfate or TiOSO4; 15% (w/v) solution in diluted H2SO4; Cat. No. 49,537-9] and the 4-amino-3-hydroxy-1-naphtelenesulphonic acid (aminonaphtosulphonic Acid; 98+% ACS reagent; Cat. No. 39,896-9). D-Sorbitol (high purity; Cat. No. SO0850) is from Scharlau. Tris (for analysis; Cat. No. 1.08382), hydrochloric acid fuming (37%; Cat. No. 1.00317) and 2-mercaptoethanol (for synthesis; Cat. No. 8.05740) are from Merck. BSA fraction V, fatty acid free (Cat. No. 775 827), adenosine-5'- diphosphate disodium salt (ADP; Cat. No. 127 507), cytochrome c (from horse heart, salt free, Cat. No. 103 888), and Triton X-100 (especially purified for membrane research; Cat. No. 789 704) are from Roche. For protein determination, the Bio-Rad protein assay dye reagent concentrate (450ml; Cat. No. 500-0006) is used. Phosphate-buffered saline (PBS 1×, liquid, pH 7.4 ± 0.05; Cat. No. 10010) is from GIBCO (Invitrogen Corporation).
Elvejhem-type glass homogenisers with Teflon pestles, as well as the Dounce-type glass potter, are from local glassware providers (VidraFoc and Sumalsa). Fifty-milliliter polypropylene copolymer centrifuge tubes (Oak Ridge centrifuge tubes; Cat. No. 3119-0050) and the 0.2-µm syringe filters (Cat. No. 190-2520) are from Nalgene.
Centrifugations are performed in a Sorvall RC 5B Plus refrigerated centrifuge with the Sorvall HS-4 swinging rotor and the SS-34 fixed angle rotor. An Eppendorf 5415 C microfuge kept in the cold room is used to centrifuge the Eppendorf tubes.
Mitochondrial oxygen consumption measurements are obtained with a Hansatech CBID oxygen electrode and registered in a PC using software from Pico Technology Limited. The polytetrafluoroethylene (PTFE) membrane (ordering code: S4; thickness 0.0125mm, width 25.4mm, 30m reel) is from Hansatech.
Spectrophotometric measurements are performed in a Unicam UV 500 spectrophotometer, where the temperature is kept constant by a DBS PCD 150 water peltier system. Data from these measurements are registered by a PC using the Vision 32 version 1.25 software from Unicam Limited.
III. PROCEDURES
A. Isolation of Mitochondria
1. Solutions
- Homogenisation medium A: 0.32M sucrose, 1mM EDTA, and 10 mM Tris-HCl. For 1 liter, weigh 109.5 g of sucrose, dissolve in bidistilled water, and add 2ml of 0.5M disodium EDTA stock solution and 20ml of 0.5 M Tris-HCl, pH 7.4. Check and adjust, if necessary, pH to 7.4 and autoclave at low pressure. Store at 4°C.
- Homogenisation medium AT: 0.075M sucrose, 0.225M mannitol, 1mM EGTA, and 0.01% BSA. For 500ml, add 12.84g of sucrose, 20.5g of mannitol, and 5 ml of a stock solution of 100 mM EGTA. Adjust pH to 7.4 and autoclave at low pressure. Store at 4°C. Just before use add 500µl of 10% (w/v) BSA. Once the BSA is added, the solution cannot be autoclaved again.
- Homogenisation medium IB 10×: 0.35M Tris-HCl, pH 7.8, 0.25M NaCl, and 50mM MgCl2. To prepare 100 ml, add 70 ml of 0.5 M Tris-HCl, pH 7.8, 5 ml of 5 M NaCl, and 5 ml of 1M MgCl2. Complete to 100 ml with bidistilled water and sterilize by filtration. Store at 4°C.
- MAITE medium: 25 mM sucrose, 75 mM sorbitol, 100 mM KCl, 0.05 mM EDTA, 5 mM MgCl2, 10 mM Tris-HCl, pH 7.4, and 10mM H3PO4 (pH 7.4). For 100 ml of solution weigh 0.86 g of sucrose, 1.37 g of sorbitol, and 0.74 g of KCl, dissolve, and then add 10 µl of 0.5 M EDTA, 500 µl of 1M MgCl2, 2 ml 0.5 M Tris-HCl, pH 7.4, and 68 µl of orthophosphoric acid. Adjust pH to 7.4 using Tris base 0.5 M. Sterilize by filtration through a 0.2-µm filter, make 10-ml aliquots, and store at 4°C.
- 0.5M EDTA: Weigh 93.1 g of disodium EDTA, dissolve, adjust the pH to 8.0, and complete the volume to 500ml with distilled water. Store at 4°C.
- 0.5 M Tris-HCl pH 7.4 or pH 7.8: For 500 ml, weigh 30.3 g of Tris and adjust pH to 7.4 or 7.8 with HCl. Store at 4°C.
- 100 mM EGTA: Prepare 100 ml of this solution by weighing 3.8g. In order to increase the solubility of EGTA, the pH has to be raised with NaOH when dissolving it. Once the EGTA is totally dissolved, complete to 100ml with bidistilled water. Store at 4°C.
- 1M MgCl2: For 100ml, weigh 2 g of magnesium chloride and dissolve. Store at 4°C.
- 5M NaCl: Weigh 29.3g of NaCl, dissolve, and adjust to 100 ml with bidistilled water.
- 10% (w/v) or 100mg/ml BSA: Weigh 0.5g of fatty acid-free BSA and dissolve in 5 ml of distilled water. Sterilize by filtration through a 0.2-µm filter and store at -20°C in 1-ml aliquots.
2. Isolation of Mitochondria from Rat Tissues
a. Mitochondria from Liver and Kidney
Use male Wistar rats weighing 200-300g. It is recommended that glassware, scissors, and metal sieves be sterilized at 160°C overnight. Autoclave plastic tubes at 1 atm for 20min.
- Previous to the sacrifice of the animals, fill 100-ml glass beakers with enough homogenisation medium A to cover the organ to be utilised, weigh, and place them on ice.
- Kill the animals, remove the organ of interest, and place it in a beaker with ice-cold homogenisation medium A (in this case liver or kidneys).
- Weigh the beaker to know the amount of tissue that has been extracted.
- Cut the tissue into small pieces with a pair of scissors.
- Sift the tissues and wash them with more homogenisation medium in order to remove blood and connective tissue. Put them back in the beaker and repeat steps 4 and 5 three or four times until the tissue is well cleaned.
- Add fresh homogenisation medium to the homogeniser in a proportion of 4 ml per gram of liver and 5 ml per gram of kidney.
- Transfer the pieces of tissue to the homogeniser.
- Homogenise with four up-and-down strokes in the Elvehjem-Potter with the motor-driven Teflon pestle at 600rpm.
- Transfer the homogenised tissue into the 50-ml sterile centrifuge tubes.
- Centrifuge in the HS-4 swinging rotor at 1000g (3000rpm) for 5 min at 4°C in order to pellet unbroken tissue, cells, and nuclei.
- Place eight Eppendorf tubes in a container with ice and fill them with supernatant from the previous step (this and the following steps are done in a cold room).
- Centrifuge the Eppendorf tubes for 2min at 13,000rpm in a microfuge.
- Using a sterile Pasteur pipette, remove the supernatant trying to draw out all the liquid and part of the light coloured fluffy layer on the top of the pellet without disturbing the darker part where mitochondria are.
- To wash the mitochondrial fraction, add homogenisation medium A, up to 1.5 ml, to four of the eight Eppendorf tubes and resuspend. Transfer the resuspended pellet of one tube to one of the tubes without medium A and resuspend.
- Centrifuge the four resulting tubes as described earlier.
- Repeat steps 15, 16, and 17 so that now two tubes are centrifuged.
- Repeat the same procedure once again until one tube, containing all the material, is left.
- When the last tube is centrifuged and the supernatant is removed, wash the pellet by resuspending it with MAITE medium.
- Centrifuge for 2min at 13,000rpm.
- Resuspend the pellets with 1 ml of MAITE medium.
When a bigger amount of sample is needed, the procedure can be started using twice as much supernatant and then finishing with the mitochondrial preparation in two Eppendorf tubes.
b. Mitochondria from Heart
- Follow the steps 1 to 7 described in Section III,A,2,a but instead of filling the beakers with homogenisation medium A, use homogenisation medium AT.
- Fill the homogeniser with 10ml of medium AT per gram of heart.
- Homogenise with six strokes in the Elvehjem- Potter with the motor-driven Teflon pestle at 600rpm.
- Transfer the homogenised tissue to a sterile 50-ml centrifuge tube.
- Centrifuge at 1000g (3000rpm) in the HS-4 swinging rotor for 5 min at 4°C.
- Transfer the supernatant from the previous centrifugation step to a clean centrifuge tube.
- Centrifuge at 12,000g (9000rpm) in the SS-34 fixed angle rotor for 10min at 4°C to obtain the crude mitochondrial fraction.
- Draw out and discard the supernatant.
- Resuspend the pellet in 5.5 ml of homogenisation medium AT.
- Transfer the mitochondrial suspension to four Eppendorf tubes for the washing steps in the cold room.
- Proceed hereafter as described for liver and kidney mitochondria, but if the mitochondrial fraction is obtained from one rat heart, the final fraction has to be resuspended in a smaller volume than those from liver and kidney, usually with 0.5-0.75 ml.
An alternative isolation procedure for heart mitochondria has been described previously (McKee et al., 1990) where perfusion and homogenisation of the hearts using subtilisin (0.4mg/ml) are proposed to improve the yield, the respiratory performance, and the protein synthesis activity of the isolated organelles. In our hands, the addition of subtilisin to the homogenisation medium provides a higher yield in the preparations and increases the in organello transcription rate. However, no significant improvement in the rate of incorporation of a radioactive amino acid into the mitochondrial translation products could be observed. However, the electrophoretic pattern of in organello synthesized products reveals an abnormal accumulation of low molecular weight peptides when subtilisin is used, probably due to residual peptidase activity of the subtilisin after breaking the organelles.
c. Mitochondria from Brain
- Again follow the same first five steps as for liver, kidney, and heart. Use homogenisation medium AT.
- Add 5 ml of medium AT per gram of brain to the homogeniser.
- Homogenise the pieces of brain with 10 to 15 strokes using a Dounce-type glass homogeniser with a manually driven glass pestle.
- Transfer the homogenised tissue to a sterile 50-ml centrifuge tube.
- Centrifuge at 1000g in swinging rotor for 5 min at 4°C.
- Transfer the supernatant from the previous centrifugation step to a clean sterile centrifuge tube.
- Resuspend the pellet resulting from the previous centrifugation in another 5 ml of medium AT per gram of starting tissue.
- Rehomogenise the nuclear pellet by repeating 10 to 15 strokes in the Dounce potter.
- Transfer the homogenised nuclear pellet to the same 50-ml tube where the first centrifugation was done.
- Centrifuge at 1000g in the swinging rotor for 5 min at 4°C.
- Remove this second supernatant and pour it into the tube with the first supernatant.
- Centrifuge at 12,000g (9000rpm) in the fixed angle SS-34 rotor for 10min at 4°C to obtain the crude mitochondrial fraction.
- Wash and resuspend as described for heart mitochondria (steps 8-11).
- Resuspended the final pellet in 0.9 ml of MAITE medium.
A great fraction of mitochondria is lost in the first nuclear pellet in whatever tissue is used; therefore, these steps (7-11) of rehomogenisation and a second centrifugation are especially necessary when isolating brain mitochondria because the homogenisation is much gentler than for the other tissues and the yield is too low when only one homogenisation step is done. The brain mitochondrial fraction obtained this way contains free as well as synaptic mitochondria. To separate free mitochondria from synaptosomes ("pinched off" synaptic ends with the synaptic mitochondria), several methods are available (reviewed in Whittaker, 1993). Partitioning in an aqueous two-phase system is recommended (Lopez-Perez, 1994).
3. Isolation of Mitochondria from Mammalian Cultured Cells
This procedure is the modified Gaines method (Enriquez and Attardi, 1996; Fernandez-Vizarra et al., 2002; Gaines, 1996).
- Harvest exponentially growing cells by trypsinization. Use the cells contained in ten to twelve 150-mm plates that are about 80-90% confluent.
- Transfer the cells to a 50-ml Falcon tube.
- Wash twice with cold PBS by centrifuging at 600g in the clinical centrifuge for 8 min at 4°C. In the last wash, transfer the cells to a 15-ml Falcon tube and centrifuge as before.
- Place the cells in ice. Measure the volume occupied by the packed cells.
- Resuspend the cells in one-half of the packed cell volume of IB 0.1×. This is a hypotonic medium to facilitate the breakage of the cells.
- Pipette the cell suspension into the homogeniser.
- Wash the tube where the cells are with 0.1× IB using another half volume of the originally packed cells and pipette this into the homogeniser.
- Perform four to five strokes in the homogeniser with the motor-driven Teflon pestle at 600rpm.
- Immediately add 1/10 of the initial volume of packed cells of 10× IB, to make the medium isotonic.
- Transfer the homogenised cells to a 15-ml Falcon tube.
- Centrifuge at 1600g (3500rpm) for 3min at 4°C in the HS-4 swinging rotor to pellet unbroken cells, debris, and nuclei.
- Draw out the supernatant and transfer it to a clean 15-ml Falcon tube.
- Resuspend the nuclear pellet in one-half of its volume of 0.1× IB.
- Repeat steps 6 to 11.
- Collect the supernatant from this second round of homogenisation and centrifugation and add it to the first supernatant.
- Centrifuge the supernatants at 1600 g (3500 rpm) for 3 min at 4°C in the HS-4 swinging rotor to remove remaining nuclei and unbroken cells.
- Pipette the supernatant in (normally two) Eppendorf tubes placed in ice (this one and the following steps are performed in the cold room).
- Centrifuge the Eppendorf tubes at 13,000rpm in the microfuge for 1 min.
- Wash the mitochondrial pellet with homogenisation medium 1× IB, until all the material is in one tube.
- Remove supernatant and wash the pellet by resuspending it with homogenisation medium A.
- Centrifuge the Eppendorf tube at 13,000rpm in the microfuge for 1 min.
- Remove supernatant and wash the pellet using MAITE medium.
- Centrifuge the Eppendorf tubes at 13,000rpm in the microfuge for 1 min.
- Resuspend the mitochondrial pellet in the appropriate volume of MAITE medium, which is usually around 300 µl.
B. Assessment of Purity
Two different parameters have to be considered when assessing the purity of a mitochondrial fraction. One is the enrichment of the preparation in mitochondria and the other is the presence of contaminants. The enrichment in mitochondria is evaluated by measuring the activities of mitochondrial enzymes in the initial homogenate and in the final mitochondrial preparation. We usually measure two activities: the inner membrane-bound respiratory complex IV or cytochrome c oxidase (Wharton, 1967) and the mitochondrial matrix enzyme citrate synthase (Srere, 1969). Spectrophotometric measurement of individual respiratory complex activities has been reviewed previously (Birch-Machin and Turnbull, 2001; Trounce, 1996). However, to evaluate the presence and abundance of contaminants, different approaches can be proposed. The most common contaminants in a mitochondrial preparation are microsomes (mostly derived from endoplasmic reticulum), lysosomes, and peroxisomes, and their presence is monitored by the determination of specific enzyme activities present in each contaminant particle. Good examples of these activities that we have used to assess purity are glucose-6- phosphatase for endoplasmic reticulum (Morr6, 1971), acid phosphatase for lysosomes (Trouet, 1974), and catalase for peroxisomes (Baudhuin, 1974). Electron microscopy morphometric analysis is good for estimating unidentified contaminants for which no enzymatic marker is available (Enriquez et al., 1990).
1. Treatment of Samples
Total homogenate samples that are used for spectrophotometric enzymatic activity measurements must undergo a freeze-thawing treatment in order to break the cells completely and liberate the enzymes from the subcellular particles. Crude mitochondrial fractions and mitochondrial preparations do not need such treatment, they are just divided in aliquots and kept at -70°C until they are used for the spectroscopic measurements. The single freezing and thawing step is sufficient to break them.
- Use 1-ml aliquots of total homogenate that are in Eppendorf tubes.
- Prepare a water bath at a temperature of 37°C.
- Fill an appropriate container with liquid nitrogen.
- Put the samples in the liquid nitrogen until they are completely frozen (2 min).
- Immediately pass the samples to the water bath at 37°C and keep them there until they are thawed completely (5 min).
- Repeat steps 4 and 5 four or five times.
- Aliquot the homogenate in Eppendorf tubes, 50- 100 µl in each tube, and keep them at -70°C.
2. Cytochrome c Oxidase Activity (EC 1.9.3.1)
Measurements of cytochrome c oxidase activity are performed spectrophotometrically using 5 µl of sample (nondiluted total homogenate or 1/10 diluted mitochondrial samples) in a final volume of 1 ml. The decrease of absorbance at 550nm, due to the oxidation of cytochrome c, is measured for 90 s at 38°C (Wharton, 1967). Sensitivity to KCN is used to confirm that cytochrome c oxidase activity is measured.
3. Citrate Synthase Activity (EC 4.1.3.7)
To measure citrate synthase activity, use the same amount of sample as in the case of cytochrome c activity; the final reaction volume is also 1 ml. In the spectrophotometer, measure the increase of the absorbance at 412nm due to the formation of a yellow complex of free CoA with DTNB, for 90s at 30°C (Srere, 1969). The CoA is formed in the reaction of acetyl-CoA with oxalacetate to form citrate, catalysed by citrate synthase.
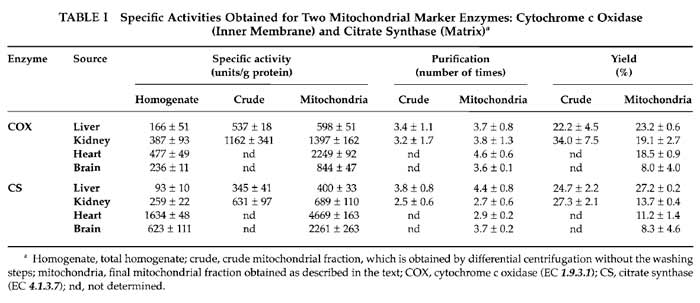
Mitochondrial enrichment assessed by these enzymatic activities in the indicated preparations is shown in Table I.
 |
4. Glucose-6-phosphatase Activity (EC 3.1.3.9)
Glucose-6-phosphatase activity was measured according to (Morré, 1971), determining spectrophotometrically the inorganic phosphate (Pi) liberated by the enzyme from the substrate (glucose-6-phosphate). The amount of sample used is 0.1ml, diluted 1/10 in the case of total homogenate and 1/20 for mitochondria and crude mitochondrial fractions. After a 15-min incubation at 37°C, there will be 0.1-1 µmol of Pi per milliliter. Protein is removed by TCA precipitation and centrifugation, and the amount of inorganic phosphate is measured in 1 ml of cleared supernatant.
5. Acid Phosphatase Activity (EC 3.1.3.2)
Incubate 1 ml of 1 / 1 0 diluted sample (total homogenate, mitochondria, or crude mitochondrial fractions) with 200 µl 0.5 M β-glycerophosphate, 100 µl of buffer, 100µl of 2% (w/v) Triton X-100, and 600µl of water for 30min at 37°C (Trouet, 1974). Remove protein by TCA precipitation and centrifugation and measure the amount of inorganic phosphate in 1 ml of cleared supernatant.
The unit of activity is defined as the amount of enzyme liberating 1 µmol of phosphate per minute.
6. Catalase Activity (EC 1.11.1.6)
Catalase activity in the samples is measured using the method described in Baudhuin (1974), which is based on the formation of the yellow titanyl sulphate- H2O2 complex. Liver and kidney samples must be diluted (1/40 for liver and 1 / 1 0 for kidney), whereas heart and brain samples do not need to be diluted. After incubation for 10min at 0°C and addition of the titanyl sulphate solution, measure the absorbance at 405 nm to evaluate how much of the initially added H2O2 is left. To calculate the activities, take into account that the reaction follows first-order kinetics and that one unit of enzyme is defined as the amount consuming 90% of the H2O2 present in a 50-ml reaction volume in 1 min.
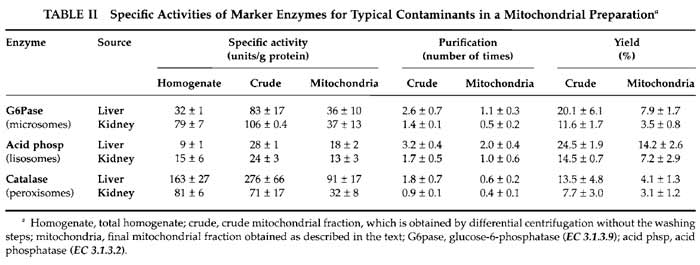
Evaluation of the presence of contaminants is shown in Table II.
 |
C. Yield of Mitochondria and Normalisation Criteria
The yield of mitochondria depends on the source of the organelles. Typically we obtain 6-9mg of mitochondrial protein per gram of starting tissue for the liver samples, which is 5-6mg/g for kidney, 2-3mg/g for heart, and 3-4.5mg of mitochondrial protein per gram of tissue in brain samples. The yield can also be calculated by the amount of mitochondrial activity (cytochrome c oxidase and citrate synthase) recovered in preparations from the total homogenate. Table I shows values obtained for the different mitochondrial preparations. The yield of mitochondria evaluated this way varies from 8% of recovery in brain to about 25% in liver.
Classically, the way of normalising mitochondrial parameters is using protein content in the sample. However, mitochondrial protein content in the different preparations is very variable. In each kind of sample, the nature and amount of contaminants vary, and even the protein composition of mitochondria is different depending on their source. In this way, specific mitochondrial enzyme activities are very different among organs; this is due to their intrinsic differences in activity and also to the different protein content in each preparation (Table I). More recently, citrate synthase activity is often taken to normalise mitochondrial parameters (Trounce, 1996), particularly respiratory chain enzyme activities, because it is considered a measurement of mitochondrial volume. However, the intertissue variation on citrate synthase specific activities is also too high and its use for normalization when comparing different sources of organelles is again questionable; i.e., when calculating the cytochrome c oxidase/citrate synthase ratio, there are significant differences between organs, with liver apparently the highest cytochrome c oxidase activity (these can easily be calculated from Table I values).
Taking all this into account, we propose that the best way to normalise mitochondrial parameters when comparing organelles from different sources is to extract mitochondrial DNA from the organelle preparations and total cell DNA from the homogenate of the same samples and quantify the amount of nuclear DNA and mitochondrial DNA. The quantification of DNA is performed easily by conventional slot blot or Southern blot and hybridisation using a specific probe for each genome (Diez-Sanchez et al., 2003). Routinely we use 12S rRNA for mtDNA and 18S rRNA for nDNA using the following rat polymerase chain reactiongenerated probes.
- 12S rRNA 617-bp amplification product (12S1 forward: caccgcggtcatacgattaacc; 12S2 reverse: ctaatttgaggagggtgacggg).
- 18S rRNA 429-bp amplification product (18S1 forward: cctcgatgctcttagctgagtg: 18S2 reverse: cagctttgcaaccatactcccc).
In this way, the parameter/nuclear DNA ratio would represent a "per cell" or "per genome" estimation and the parameter/mtDNA a "per mitochondria" or "per mitochondrial DNA" estimation. Although the mtDNA copy number per cell varies among cell types, tissues, and physiological condition, it can be monitored easily by following the mtDNA/nDNA ratio.
D. Assessment of Functionality
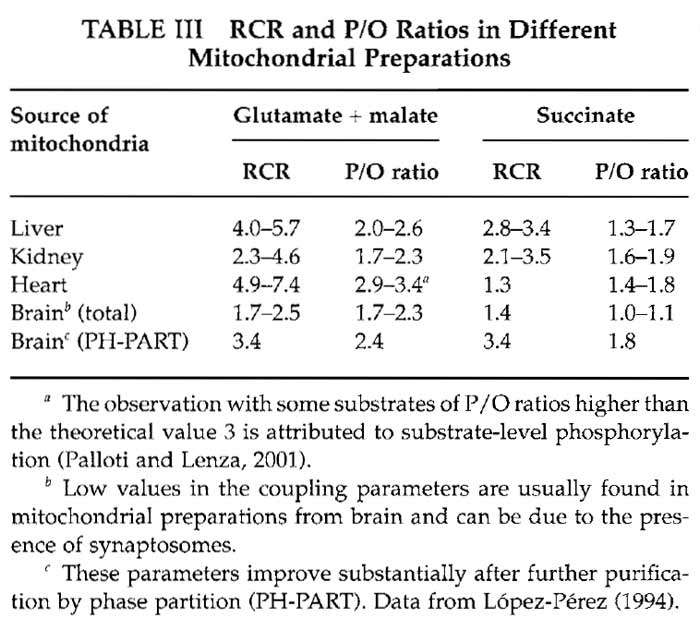
Mitochondria obtained using the purification protocols described here have intact membranes and are coupled. This means that electron transfer among the inner membrane complexes takes place when there is phosphorylation of ADP by the ATP synthetase; this is the main way to dissipate the proton gradient between the intermembrane space and the matrix. Mitochondrial electron transfer can be measured by oxygen consumption using an oxygen electrode (Trounce, 1996). The respiratory control ratio (RCR) is a way to determine how coupled mitochondria are The consumption of oxygen by mitochondria in the presence of electron donors (substrates) and ADP, called state 3 respiration, is measured and then compared with the respiration rate when all the ADP has been phosphorylated (state 4). When there is no difference between state 3 and state 4 respiration (RCR = 1), mitochondria are completely uncoupled, whereas RCRs higher than 4 are indicative of tightly coupled mitochondria. Another parameter to evaluate coupling using the same measurements in the oxygen electrode is the P/O ratio, which gives the moles of synthesized ATP per atom of oxygen transformed to water during oxidative phosphorylation. Coupled mitochondria usually exhibit P/O ratios higher than 2.5, approaching 3 with NADlinked substrates, and ratios higher than 1.8, approaching 2 with succinate (Palloti and Lenaz, 2001). A detailed explanation on oxygen electrode functioning can be found in Rickwood (1987).
- Perform measurements in the oxygen electrode using MAITE medium where mitochondria are resuspended. The final volume in the electrode chamber is 1 ml.
- Add 10µl of 100mg/ml BSA and either 20µl of 500mM glutamate and 25µl of 100mM malate when measuring RCR with glutamate and malate (NAD-linked substrates) or, as an alternative, 20µl of 500 mM succinate.
- Add 0.5 to 1 mg of mitochondrial protein from freshly isolated mitochondria. In the case of heart mitochondria, 0.2mg of protein is enough to perform the assay.
- When the chamber is closed, add 100nmol of ADP (10µl of ADP 10mM) to stimulate mitochondria and start state 3 respiration. This addition is repeated after mitochondria return to state 4 respiration.
Values for RCR and P/O for different mitochondrial preparations are given in Table III.
 |
IV. COMMENTS AND RECOMMENDATIONS
We would like to make a particular comment on the nature of the less commonly evaluated contaminants in the mitochondrial preparations, whose presence is variable depending on their source and that has been frequently underscored in classical preparative or metabolic assays. In addition to their utility in bioenergetic and metabolic studies, isolated organelles provide a unique tool to investigate the synthesis and expression of mtDNA in conditions that very much resemble the in vivo enzyme/substrate proportions, ionic composition, and integrated activity of the metabolic and biogenetic processes (Enriquez et al., 1996, 1999; Enriquez and Attardi, 1996; Fernandez-Vizarra et al., 2002).
In this type of assay the presence of contaminants can be relevant or negligible depending on the type of investigations to be performed. For example, only brain crude mitochondria preparations contain large myelin membrane debris that can be estimated by monitoring the 2', 3'-cyclic nucleotide 3'-phosphohydrolase (Enriquez et al., 1990; Olafson et al., 1969). They do not seem to interfere with biogenetic analyses, but can contribute to the overall protein content (see later). In addition, this crude preparation contains synaptosomes (also enclosing mitochondria). Crude mitochondrial preparations are suitable for in organello transcription and replication analysis when labeled UTP or dNTPs are used, as well as for protein import assays, as nucleotides or peptides do not cross the plasma membrane of synaptosomes. However, when analyzing protein synthesis and amino acylation using labeled amino acids, one should keep in mind that amino acids are also imported by synaptosomes and used by their mitochondria. Therefore, if trying to evaluate the influence of specific factors in the mitochondrial protein synthesis, one has to be aware that a relevant portion of mitochondria are protected by a plasma membrane.
Other relevant contaminants, not considered very often, are residual but partially active biogenetic components of the nucleocytoplasmic machinery, such as cytoplasmic ribosomes, transcriptionally or replicative partially active nuclear rests, or cytoplasmic aminoacyl tRNA synthetases. The use of additional purification steps using density gradient purification could reduce the presence of some of these contaminants, but it is expensive, not suitable for large-scale preparations, and, most important, can affect the functionality of the purified organelles. Protein synthesis due to contaminant cytoplasmic ribosomes is not very relevant (Fernandez-Vizarra et al., 2002) and it is possible to eliminate it completely using drugs that specifically inhibit cytoplasmic protein synthesis without affecting mitoribosomes such as cycloheximide or emetine. In our hands, no detectable aminoacylation of cytoplasmic tRNAs is observed when using purified organelles from cultured cells (Enriquez, 1996). The same is true for transcription in any source of mitochondria tested (Andreu et al., 1998; Enríquez, 1999; Enriquez et al., 1991, 1996; Fernandez-Vizarra et al., 2002; Micol et al., 1997), but a background of nonmitochondrial DNA is labeled when mitochondria are prepared from exponential growing cultured cells (unpublished results). To avoid this, it is recommended to add DNase (or microccocal nuclease) during the mtDNA replication assay, as mtDNA and mtRNAs are protected by the mitochondrial double membrane. Then, as the standard DNA isolation procedure, after incubations, includes a strong step of proteinase K digestion, the DNase is fully removed before breaking of the mitochondria. In that way only mitochondrial DNA is labeled.
V. PITFALLS
- Always keep mitochondrial preparations on ice while they are being isolated and until they are used for biogenetical analyses or for measurements in the oxygen electrode.
- Invert the tubes where the mitochondria are kept every once in a while in order to maintain mitochondria in suspension, avoiding sedimentation. During the incubation for activity determination, mixing is essential, as sedimentation of the organelles can make them become permanently impaired in their biogenetic activities due to a lack of oxygenation.
- Irreversible uncoupling of mitochondria during purification can occur and is found more often when isolating brain or heart mitochondria. This is likely due to the well-known uncoupling activity of fatty acids and could be prevented by the inclusion of fatty acidfree BSA in the purification buffer.
Acknowledgments
We thank Drs. Julio Montoya, Acisclo Pérez-Martos, and Manuel, José López-Pérez for their valuable input in our work and Santiago Morales for his technical assistance. Our research was supported by the Spanish Ministry of Education PM-99-0082 grant to JAE, by the Ramón y Cajal 2001 grant to PF-S, and by a Diputación General de Arag6n (CONSID B015/2001) fellowship to EF-V.
References
Andreu, A. L., Arbos, M. A., Perez-Martos, A., Lopez-Perez, M. J., Asin, J., Lopez, N., Montoya, J., and Schwartz, S. (1998). Reduced mitochondrial DNA transcription in senescent rat heart. Biochem. Biophys. Res. Commun. 252, 577-581.
Baudhuin, P. (1974). Isolation of rat liver peroxisomes. Methods Enzymol. 31, 356-368.
Birch-Machin, M. A., and Turnbull, D. M. (2001). Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 65, 97-117.
Diez-Sanchez, C., Ruiz-Pesini, E., Lapena, A. C., Montoya, J., Perez- Martos, A., Enriquez, J. A., and Lopez-Perez, M. J. (2003). Mitochondrial DNA content of human spermatozoa. Biol. Reprod. 68, 180-185.
Enríquez, J. A., and Attardi, G. (1996). Analysis of Aminoacylation of Human Mitochondrial tRNAs. Methods Enzymol. 264, 183-196.
Enríquez, J. A., Fernandez-Silva, P., Garrido-P6rez, N., L6pez-P6rez, M. J., P6rez-Martos, A., and Montoya, J. (1999). Direct regulation of mitochondrial RNA synthesis by thyroid hormone. Mol. Cell Biol. 19, 657-670.
Enriquez, J. A., Lopez-Perez, M. J., and Montoya, J. (1991). Saturation of the processing of newly synthesized rRNA in isolated brain mitochondria. FEBS Lett. 280, 32-36.
Enriquez, J. A., Perez-Martos, A., Lopez-Perez, M. J., and Montoya, J. (1996). In organello RNA synthesis system from mammalian liver and brain. Methods Enzymol. 264, 50-57.
Enriquez, J. A., Sanchez-Prieto, J., Muino Blanco, M. T., Hernandez- Yago, J., and Lopez-Perez, M. J. (1990). Rat brain synaptosomes prepared by phase partition. J. Neurochem. 55, 1841-1849.
Fernandez-Vizarra, E., Lopez-Perez, M. J., and Enriquez, J. A. (2002). Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods 26, 292-297.
Gaines, G. L., 3rd (1996). In organello RNA synthesis system from HeLa cells. Methods Enzymol. 264, 43-49.
Lopez-Perez, M. J. (1994). Preparation of synaptosomes and mitochondria from mammalian brain. Methods Enzymol. 228, 403-411.
McKee, E. E., Grier, B. L., Thompson, G. S., and McCourt, J. D. (1990). Isolation and incubation conditions to study heart mitochondrial protein synthesis. Am. J. Physiol. 258, E492-E502.
Micol, V., Fernandez-Silva, P., and Attardi, G. (1997). Functional analysis of in vivo and in organello footprinting of HeLa cell mitochondrial DNA in relationship to ATP and ethidium bromide effects on transcription. J. Biol. Chem. 272, 18896-18904.
Morré, D. J. (1971). Isolation of Golgi apparatus. Methods Enzymol. 31, 130-148.
Olafson, R. W., Drummond, G. I., and Lee, J. F. (1969). Studies on 2',3'-cyclic nucleotide-3'-phosphohydrolase from brain. Can. J. Biochem. 47, 961-966.
Palloti, E, and Lenaz, G. (2001). Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol. 65, 1-35.
Rickwood, D., Wilson, M. T., and Darley-Usmar, V. M. (1987). Isolation and characteristics of intact mitochondria. In "Mitochondria: A Practical Approach" (V. M. Darley-Usmar, D. Rickwood, and M. T. Wilson, eds.) pp. 1-16. IRL Press, Oxford.
Srere, P. A. (1969). Citrate synthase. Methods Enzymol. 13, 3-11.
Trouet, A. (1974). Isolation of modified liver lysosomes. Methods Enzymol. 31, 323-329.
Trounce, I. A., Kim, Y. L., Jun, A. S., and Wallace, D. C. (1996). Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 264, 484-509.
Wharton, D. C., and Tzagoloff, A. (1967). Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 10, 245-250.
Whittaker, V. P. (1993). Thirty years of synaptosome research. J. Neurocytol. 22, 735-742.
Support our developers




