Methods in Apoptosis
I. INTRODUCTIONApoptosis and necrosis are two mechanisms of cell death, each with its own distinguishing morphological and biochemical features. Necrosis, which occurs within seconds of cell insult (Majno and Joris, 1995), may be described as "cell murder" resulting from external damage to the cell membrane, loss of homeostasis, water and extracellular ion influx, intracellular organelle swelling, cell rupture (lysis), and so inflammatory cell attraction. Initially described by Kerr et al. (1972), apoptosis is a much slower process of events than necrosis, requiring from a few hours to several days (depending on the initiator) and resulting from molecular signals initiated within individual cells (see Nagata, 1997; Barinaga, 1998; Van Cruchten and Van Den Broeck, 2002). The initiators of apoptosis that instigate the cascade of events leading to activation of a series of cytoplasmic proteases, termed caspases (cysteinyl-asparatate-specific proteinases), are multiple. Two such pathways involve (i) activation of cell surface death receptors, resulting in direct activation of caspases, and (ii) cytochrome c release from the mitochondria into the cytoplasm following induction of leakiness in its membrane. The terminal caspases downstream from these initiator mechanisms lead to the morphological and biochemical events of apoptosis.
The mechanisms of apoptosis, which is analogous to "cell suicide," are essentially the same, whether induced by genetic signals or through external initiators. The events involved include cell membrane blebbing; chromatin aggregation; nuclear and cytoplasmic condensation leading to cell shrinkage; and partitioning of cytoplasm and nucleus into membrane-bound apoptotic bodies, which contain ribosomes, morphologically intact mitochondria, and nuclear material. As a result of the efficient mechanism for the removal of apoptotic cells by phagocytic cells in vivo, an inflammatory response is not stimulated. In vitro apoptotic bodies swell and finally lyse (Darzynkiewicz and Traganos, 1998; Kiechle and Zhang, 2002).
Apoptosis is key to many fundamental aspects of biology, including embryonic development and normal tissue homeostasis, as well as in many pathological events, such as loss of regulated cell death in cancer, response of cancer cells to chemo- and radiotherapy (Clynes et al., 1998), and death of cells in diabetes (Sesti, 2002) and neurodegenerative diseases (Vila and Przedborski, 2003). Accurate detection of apoptosis is of great importance to increase our understanding of biological events that may allow us to understand and to manipulate these events as a form of therapy.
Major elements involved in the apoptotic pathway, which should be considered when selecting suitable methods for apoptosis detection, include the following.
- Death receptor activation: Following receptor cross-linking by ligand (e.g., the Fas receptor by the CD95 (APO-1 Fas) or Tumor Necrosis Factor (TNF) receptor type 1 by TNF), signal transduction leads to caspase activation (see (f) below).
- Changes in cellular morphology: As described earlier.
- Membrane alterations: Translocation of phosphatidylserine (PS) from the cytoplasmic to the extracellular side of the cell membrane is an early event in apoptosis.
- DNA fragmentation: Prior to the induction of cell membrane permeability, fragmentation of genomic DNA at sites located between nucleosomal units, generating mono- and oligonucleosomal DNA fragments, irreversibly commits the cell to die.
- Disruption of mitochondria: As described, disruption of the mitochondrial membrane results in cytochrome c (Apaf-2) release. This subsequently promotes caspase activation by binding to Apaf-1 and inducing activation of Apaf-3 (caspase-9). Similarly, release of apoptosis-inducing factor (AIF) induces apoptosis.
- Activation of caspases: At least 11 different caspases have been identified in mammalian cells. Activation of this protease cascade via a range of stimuli is central to the execution of apoptosis.
The range of techniques and methods for analysis of apoptosis is extensive. Due to space limitations in this article, we do not propose to describe all methods comprehensively. Table I lists a number of techniques for analysis of the cellular events described earlier. A selection of techniques used for studying these events is described in further detail.
 |
II. MATERIALS AND INSTRUMENTATION
A. Light Microscopy (LM) and Fluorescence Microscopy (FM)
Frost-ended slides and coverslips (Chance Propper); ice-cold methanol; Coplin jars; forceps; micropipettes; grease pen (DAKO S2002); and mounting medium (Vectashield mounting solution with antifade additive [Vector Labs.; H-1000]) suitable for fluorescence slides and may also be used for LM slides. Alternatively, 20% glycerol prepared in H2O is also suitable for mounting slides for LM.
If analysing suspension cells, a cytospin (e.g., Heraeus Labofuge 400) and cytospin cups are required.
For LM only: haematoxylin, aluminium potassium sulphate, citric acid, and chloral hydrate.
For FM only: Stains include 4',6-diamidino-2- phenylindole (DAPI, Sigma D-9542), propidium iodide (PI, Sigma P-4170), Hoechst 33258 (Sigma B-2883), Hoechst 33342 (Sigma B-2261), and acridine orange (AO, Sigma A-6014) in phosphate-buffered saline (PBS), pH 7.4.
B. Gel Electrophoresis for DNA Ladder
Horizontal agarose gel electrophoresis chamber and combs (Bio-Rad); electric power supply; UV transilluminator or gel analyser (e.g., EpiChemi II Darkroom, UVP Laboratory Products); PBS (Oxoid BR14a); ethidium bromide (Sigma E-8751); agarose (Sigma A-9539); Tris (Sigma T-8524); EDTA (Tris E-5134); NaCl (Sigma S-9899); sodium dodecylsulfate (SDS) (BDH 442152V); RNase A (Sigma R-5250); proteinase K (Sigma P-2308); boric acid (Sigma B-7901); bromphenol blue (Sigma B- 5525); glycerol (Sigma G-2025); molecular markers, e.g., Phi X174 DNA HaeIII digest (Sigma D-0672); micropipettes.
C. Terminal Deoxynucleotidyl Transferase- Mediated Deoxyuridine Triphosphate Nick End-Labelling Assay (TUNEL)
Apoptosis detection system: (1) fluoresceincontaining equilibrating buffer, (2) nucleotide mix, (3) TdT enzyme, (4) 20X SSC solution, (5) proteinase K, (6) protocol (Promega; G3250); plastic coverslips, glass slides and coverslips (Chance Propper); propidium iodide (Sigma P-4170); Coplin jars; forceps; humidifying chamber; 37°C incubator; Triton X-100 (Sigma T- 8787), PBS (Oxoid BR14a); 4% paraformaldehyde (Sigma P-6148) in PBS (pH 7.4) (freshly prepared); Vectashield mounting solution with antifade additive (Vector Labs.; H-1000); 70% ethanol [prepare from absolute ethanol (Sigma E-7037)]; and micropipettes.
Note: Items 1-6 are included in the apoptosis detection system available commercially from Promega (G3250). There are, however, other detection kits available commercially that may be equally suitable.
D. Reverse Transcriptase-Polymerase Chain Reaction
As all general laboratory glassware, spatulas, etc., are often contaminated by RNases, these items should be treated by baking at 180~ for a minimum of 8h. Sterile, disposable plasticware is essentially free from RNases and so generally does not require pretreatment. All solutions/buffers used should be prepared in baked glassware using sterile ultrapure water treated by the addition of diethylpyrocarbonate (DEPC) [Sigma D-5758, (0.1%, v/v)] and autoclaved. As for all laboratory procedures described in this article, gloves should be worn at all times to protect both the operator and the experiment. This, too, prevents the introduction of RNases and foreign RNA or DNA in the reverse transcriptase (RT) and polymerase chain reaction (PCR).
1. For RNA Isolation and Quantification
TRI Reagent (Sigma T-9424), chloroform (Sigma C-2432), isopropanol (Sigma 1-9516), ethanol [Sigma E-7037; prepare as 75% (v/v) in H2O], DEPC, micropipettors, tips, Eppendorf tubes, etc., spectrophotometer (e.g., SpectraMax Plus plate reader, Molecular Devices), and quartz cuvettes or Nanodrop (ND-1000; Labtech Int. Ltd.)
2. For RT and PCR Reactions
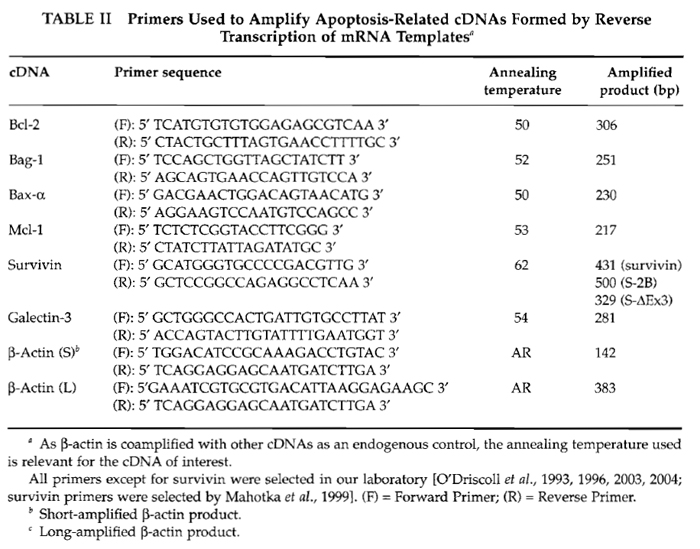
DEPC-treated H2O; oligo(dT)12-18mer (Oswel, Southampton, UK); MMLV-RT enzyme (200U/µl) (Sigma M-1302); 5X buffer (Sigma B-0175); dithiothreitol (DTT, 100mM) (Sigma D-6059); RNasin (40U/ml) (Sigma R-2520); dNTPs (10mM each of dATP, dCTP, dGTP, and dTTP for RT; 1.25 mM each for PCR) (Sigma DNTP-100); MgCl2 (25 mM) (Sigma M-8787), Taq DNA polymerase enzyme (5U/µl) (Sigma D-6677); 10X buffer (Sigma P-2317); target primers and internal control primer (see Table II) (for further details on primer selection criteria, see O'Driscoll et al., 1993); thermocycler.
 |
3. Gel Electrophoresis
Amplified products are analysed by gel electrophoresis (see Section IIIB).
Note: Several of the stains and other reagents used (e.g., DEPC) should be handled with caution as they are known or thought to be toxic, carcinogenic, and/or mutagenic.
III. PROCEDURES
A. Light and Fluorescence Microscopy Detection of Apoptotic Cell Morphology
As described previously, a cell undergoing apoptosis proceeds through various stages of morphological change (Wilson and Potten, 1999). Light microscopy and fluorescence microscopy are probably the simplest and most basic techniques by which such apoptotic cell death can be investigated. A broad range of stains and dyes are available to assist in the assessment of nuclear morphology. For light microscopy, the nuclear stain haematoxylin is used frequently (often with eosin as a counterstain). The most commonly used DNA nucleic acid-reactive fluorochromes for UV light fluorescence microscopic analysis of fixed (porated using, e.g., methanol or ethanol) cells include DAPI, PI, Hoechst 33258 and 33342, and AO. (As mentioned previously, PI, DAPI, and Hoechst are also very useful for assessing the membrane permeability of cells).
Using LM, cells dying by apoptosis are identified by their reduced size, cell membrane "blebbing/ budding," and loss of normal nuclear structural features- nuclear fragmentation and chromatin condensation. In contrast, characteristic features of necrotic cell death include cell and nuclear swelling, cytoplasmic vacuolisation, patchy chromatin condensation, and plasma membrane rupture.
If assessing adherent cell types, the cells may be grown directly on coverslips (plated in Petri dishes). For suspension cells, cytospins are generally prepared. For both monolayer and suspension cultures, it is important to consider cell concentration. Fifty to 70% confluency of the area being analysed is generally considered optimalmif the cells are more confluent, overlapping may occur, which may hinder analysis; if cells are too sparse, an accurate examination may not be achievable. Typically, 1-2 × 105 cells suspended in 100µl PBS containing 1% (w/v) fetal calf serum (FCS) should be cytocentrifuged onto microscope slides using a low-speed, short centrifugation (e.g., 600- 1000rpm for 2-4min); however, this should be optimised as relevant for cells being analysed.
Solutions
For LM only. 0.1M haematoxylin: In a fume hood, dissolve 1 g haematoxylin (BDH 34242) in 1 litre distilled water, boil for 5 min, remove from heat, and add 0.2 g sodium iodate (Sigma S-4007). After 10 min, in the order listed, add 50g aluminium potassium sulphate (Sigma A-7167), 1 g citric acid (Sigma C-2404), and 50g chloral hydrate (Sigma C-8383), allowing each to dissolve completely prior to adding the next. The complete solution is stable for approximately 3 months at room temperature.
For FM only. Fluorescent stains should be stored in light-proof containers to prevent quenching.
- DAPI: Prepare stock at 5 mg/ml in methanol (store at -20°C for up to 3 months). Prior to use, dilute 1:10,000 in PBS, pH 7.4.
- PI: Prepare stock at 2mg/ml in PBS, pH 7.4 (store at -20°C for up to 3 months). Prior to use, dilute 1:1500 in PBS.
- Hoechst 33258 and 33342: Prepare stock at 100µg/ml in PBS, pH 7.4 (store at -20°C for up to 3 months). Prior to use, dilute 1:10 in PBS.
- AO: Prepare stock at 2mg/ml in PBS, pH 7.4 (store at -20°C for up to 3 months). Prior to use, dilute 1:400 in P BS.
Steps
Note: When working with fluorochromes, minimise exposure to light at all stages of preparation.
- Fix cells in ice-cold methanol for 5-7 min.
- Allow to air dry.
- Using a grease pen, encircle area of cells for analysis. Typically a circle of 10 mm diameter or smaller is drawn (to contain solutions and to minimise volumes of solutions, antibodies, etc., required).
- Stain cells with 0.1% hematoxylin (for LM) or DAPI, PI, Hoechst, or AO (for FM) for 3-5 min.
- Wash in distilled H2O (for LM) or PBS (for FM).
- Mount coverslip on slide using an aqueous-based mountant medium and fix in place (clear nail varnish may be used to secure coverslip).
- Immediately analyse and photograph samples by fluorescence microscopy. Slides cannot be stored long term as fluorescence quenches. However, storing at 4°C in the dark will prolong the life span of the signal. Haematoxylin-stained slides may be stored indefinitely.
B. Gel Electrophoresis for DNA Ladder
Activation of endogenous Ca2+- and Mg2+- dependent nuclear endonuclease(s) cleaves DNA into discrete fragments, initially into 300- to 50-kb fragments and subsequently into 180-bp fragments. In brief, for DNA fragment detection by gel electrophoresis, DNA is extracted from cells and loaded onto a 1.5% agarose gel containing ethidium bromide. The DNA fragments form a characteristic "ladder" pattern resulting from multiples of a 180-bp DNA subunit, representing DNA of the size of individual nucleosomes and oligonucleosomes. [Nuclear DNA damage resulting in death by necrosis, however, is random and results in smears on a gel (Ramachandra and Studzinski, 1995).]
Solutions
- Lysis solution: 50mM Tris-HCl (pH 8.0), 20mM EDTA, 10mM NaCl, and 1% (w/v) SDS. A 50-ml aliquot stock may be prepared and aliquotted in 2-ml volumes (store at -20°C for up to 3 months).
- TBE buffer: 1X TBE consists of 10.8g Tris base, 5.5 g boric acid, and 4 ml 0.5 M EDTA (pH 8.0) made up to 1 litre with ultrapure water. It is advisable to prepare as a 10X concentrate and store at room temperature for up to 3 months.
- RNase A: Prepare stock of 200µg/ml and store at -20°C (for up to 3 months) to use at a final concentration of 20µg/ml.
- Proteinase K: Prepare stock of 1 mg/ml and store at -20°C (for up to 3 months) to use at a final concentration of 100µg/ml.
- Ethidium bromide: Prepare a 10 mg/ml stock of ethidium bromide by adding 1 g of ethidium bromide to 100 ml of H2O. Stir on a magnetic stirrer for several hours to ensure that the dye has dissolved. Wrap the container in aluminium foil or keep in a dark bottle. Store at room temperature. Note: It is important to consider that ethidium bromide is a potential carcinogen. Due caution should be taken when preparing ethidium bromide stock, adding ethidium bromide to gel, disposing of exposed pipette tips and ethidium bromidecontaining gels, etc.
- DNA loading buffer: DNA loading buffer may be prepared at a 6X concentrate by mixing 1 mg/ml bromphenol blue, 1 mM EDTA, and 50% glycerol (v/v) in ultrapure H2O. Store at room temperature (should be stable for up to 12 months).
Steps
- Harvest cells for analysis, wash with PBS, and pellet by centrifugating for 5 min at 1000rpm.
- Lyse cell pellet by adding a volume of lysis solution containing 50mM Tris-HCl (pH 8.0), 20mM EDTA, and 10mM NaCl, 1% (w/v) SDS and incubating for 10min at 37°C (the cell number used and the volume of lysis solution added must be optimised for each cell type being analysed because if the sample is too viscous the resulting gel resolution may be reduced). It is important to ensure at this stage that the pellet has broken up. This may be assisted by "flicking" the tube gently.
- Add RNase A (final concentration of 20µg/ml) to the lysate and incubate for 60min at 37°C.
- Add proteinase K (final concentration of 100µg/ml) to the lysate and incubate for 4h at 37°C. Lysates may then be loaded directly onto an agarose gel (as described later). If clear bands are not detected using whole cell lysates, DNA may be purified from cell lysates at this stage using standard phenol/chloroform/ isoamyl alcohol procedures (see Sambrook and Russell, 2001).
- During the incubation steps, dilute a stock of 10X TBE to 1X TBE to use in preparation of a 1.5% agarose gel. 1X TBE buffer is also prepared for use as running buffer.
- Melt and pour 1.5% agarose gel in a horizontal gel support (taking safety pre cautions when working with molten agarose). Insert the comb and allow the gel to solidify.
- Add loading buffer to lysates (to resulting in a final 1X concentration of loading buffer), "flick" tubes, and centrifuge briefly.
- Load 10- to 50-µl samples to the gel wells and run the gel in 1X TBE at 75V for approximately 1.5h at room temperature. Molecular weight markers should be loaded to allow lysate band sizes to be estimated following electrophoresis.
- Stain gel with 5µg/ml ethidium bromide in 1X TBE for 30min.
- Place the gel on a UV transilluminator box to visualise (and photograph) resolved DNA fragments in a ladder pattern. (Note: Wear appropriate safety gear to ensure that eyes and skin are not exposed to UV radiation.)
C. TUNEL
TUNEL is a cytochemical method suitable for analysis of apoptotic DNA fragmentation in individual cells. This technique involves in situ enzymatic labelling of the 3'-OH ends of fragmented DNA in fixed, permeabilised cells with either enzyme (phosphatase or peroxidase) or fluorochrome-tagged deoxynucleotides using terminal deoxynucleotidyl transferase (TdT). (Note: If DNA polymerase I is used instead of TdT, the method is termed in situ nick translation.) If fluorescence labelling is chosen, the fluorescein-12-dUTP-labelled DNA can then be visualised directly by fluorescence microscopy to give qualitative results. If facilities are available and quantitative results are required, flow cytometric analysis may be used.
Solutions
- Fixation solution: 4% paraformaldehyde in PBS (pH 7.4) is prepared immediately before use. To help dissolve the paraformaldehyde, this solution may be placed on a magnetic stirring box and heated very gently. Paraformalydehyde should only be prepared and used in a fume hood.
- Permeabilisation solution: 0.1% Triton X-100 in PBS.
Steps
- Grow cells for analysis as a monolayer on glass slides. Alternatively, cytospin preparations of cells may be used.
- Remove culture medium, wash cells twice with PBS, and allow to air dry.
- Fix cells by immersing slides in a Coplin jar containing fixation solution, i.e., freshly prepared 4% paraformaldehyde in PBS (pH 7.4), for 25 min at 4°C
- Rinse the slides by immersing in PBS for 5 min at room temperature (two times). It is very important that the slides are not allowed to dry out during the following steps.
- Gently tap off excess PBS, cover cells with 100 btl of equilibration buffer, and incubate for 5-10min at room temperature.
- While incubating in equilibration buffer (step 5), prepare the TdT incubation buffer by adding 45µl equilibration buffer + 5µl nucleotide mix + 1 µl TdT enzyme per sample (prepare as a master mix, depending on the number of samples being analysed). As a negative control, TdT may be eliminated from some samples and replaced with an equal volume of ultrapure H2O. It is important to keep the nucleotide mix and TdT incubation buffer on ice and to ensure that all steps from now on be protected from direct light.
- Following equilibration, carefully blot off excess equilibration buffer and add 50µl of TdT incubation buffer to the cells. Cover with plastic coverslips to ensure even distribution of the TdT incubation buffer and to prevent slides from drying out.
- Place slides in a humidified chamber (a suitably sized flat-bottomed plastic box containing a layer of paper towels soaked in water is ideal for this purpose). Cover with aluminium foil to protect from light and incubate for 60min at 37°C to allow the "tailing" reaction to occur.
- To terminate the reaction, 20X SCC buffer should be diluted to a 2X SCC solution with ultrapure water and placed in a Coplin jar, the coverslips removed from the slides, and the slides immersed for 15 min at room temperature.
- To remove unincorporated fluorescein-12- dUTP, the slides should be washed by placing in PBS for 5min at room temperature (repeat wash two times).
- If desired, slides may be stained at this stage by immersing in Coplin jars containing freshly prepared PI solution (see preparation details given earlier) and incubating for 15 min at room temperature. Following this, wash slides in ultrapure water (3 × 5 min at room temperature).
- Mount slides using Vectashield and glass coverslips.
- Immediately analyse and photograph cells by fluorescence microscopy (520 mm filter for fluorescein and >620 mm filter if propidium iodide has been included. Slides cannot be stored long term as fluorescence quenches. However, storing at 4°C in the dark may prolong the life span of the signal.
D. RT.PCR
Expression of apoptosis-related mRNAs may be analysed using RT-PCR methods. This section describes the use of "basic" RT-PCR, but it is important to be aware that depending on the requirements of the study and the resources available, RT-PCR may be used to indicate the presence or absence of transcripts of interest or it may be developed as a semiquantitative or a quantitative level using real-time PCR.
RNA may be isolated from cells using the procedure described by Chomczynski and Sacchi (1987). However, there are now a number of less laborious, commercially available methods, including the use of TRI reagent (Sigma), as described here. The RT procedure detailed involves use of the MMLV-RT (Sigma) enzyme for cDNA synthesis and the PCR uses Taq DNA polymerase (Sigma). Again, however, there are other reverse transcriptases and DNA polymerase enzymes available that may be equally suitable.
Primers suitable for the analysis of human bcl-2, bag-l, bax-α, mcl-1, galectin-3 (designed in our laboratory), and survivin are included in Table II, as examples of apoptosis-related gene transcripts that may be desirable to analyse by RT-PCR. There are, of course, many more mRNAs involved in apoptotic pathways that should be considered. The methods described here may be adapted by using relevant primers for the amplification of other apoptosis-related cDNAs [for information on primer design and selection criteria, see O'Driscoll et al. (1993)].
Table II also includes primer sequences for coamplification of cDNA derived from an endogenous "housekeeping" mRNA, β-actin, as control. Inclusion of such primers serves to indicate that the RT and PCR reactions have been performed successfully; if performing semiquantitative PCR, this allows the apoptosis-related gene transcript results to be normalised relative to a control that (generally) should be constant.
Steps
RNA Isolation and Quantitation
- Pellet cells and wash with PBS (three times).
- In a 0.5-ml Eppendorf tube, lyse cells in TRI reagent; 1 ml TRI reagent is suitable to lyse approximately 0.5-1 × 107 cells.
- RNA may be isolated immediately or lysates stored at -80°C for up to 1 month.
- Incubate lysates at room temperature for 5 min.
- Add 200 µl chloroform per 1 ml TRI reagent used, shake samples vigorously for 15s, and incubate at room temperature for 15 min.
- Centrifuge at 12,000g, at 4°C for 15 min. Following this, the RNA will be contained in the upper aqueous phase, below which are the DNA-containing interface and the protein-containing organic phase.
- Transfer the RNA-containing aqueous phase to a clean 0.5-ml Eppendorf tube, add 0.5ml isopropanol, mix, and incubate at room temperature for 10 min.
- Centrifuge at 12,000g at 4°C for 10min to pellet RNA.
- Remove supernatant (carefully, to prevent disturbing RNA pellet), wash RNA with 1 ml 75% ethanol, vortex for 5s, and centrifuge at 7500g at 4°C for 5 min.
- Remove ethanol, air dry pellet briefly, and resuspend in 25-50µl DEPC-H2O. (Ensure that the pellet does not dry completely, as this decreases its solubility greatly. The solubility of RNA can be improved by heating to 55-60°C with intermittent vortexing or by passing the RNA through a pipette tip, if necessary). Store RNA at -80°C.
- Quantify RNA spectrophotometrically at 260 nm and 280nm. The A260/A280 ratio of RNA is approximately 2. [Partially solubilised RNA has a ratio <1.6 (Ausubel et al., 1991).]
Typical RT Reaction
- To a 0.5-ml Eppendorf tube add 1µl oligo(dT)12-18mer (1 µg/µl), 1 µl RNA (at 1 µg/µl), and 3µl DEPC-H2O. Mix gently, incubate at 70°C for 10 min, and chill on ice. [Note: For survivin analysis, to avoid coamplification of the homologous cDNA for effector protease receptor-1 (EPR-1), instead of including the oligo(dT) primer, use 250ng of the survivinspecific RT primer 5' AGGAACCTGCAGCTCAGA 3'.]
- Add the following reagents: 4µl 5X buffer, 2µl (100 mM) DTT, 1 µl (40 U / µl) RNasin, 1 µl (10 mM each) dNTPs, and 6 µl (200 U/µl) MMLV-RT. Mix.
- Incubate at 37°C for 1 h, followed by 95°C for 2 min.
Typical PCR Reaction
- To a 0.5-ml Eppendorf tube add the following reagents: 24.5 µl H2O, 5 µl 10X buffer, 3 µl (25 mM) MgCl2, 8µl (1.25mM) dNTPs, 1µl target forward primer (250ng/µl) (see Table II), 1µl target reverse primer (250ng/µl), 1µl control forward primer (250ng/µl), 1µl control reverse primer (250ng/µl), and 0.5 µl (5 U/µl) Taq polymerase enzyme. Mix gently. Add 5µl cDNA (from RT reaction). Mix gently and centrifuge very briefly to collect solution at bottom of tube. (Note: Primer concentrations used may need to be increased or decreased to optimise amplification.)
- Amplify on a thermocycler using the following PCR cycle: 94°C for 2min; 30 cycles of 94°C for 30s, relevant annealing temperature (Table II) for 30 s, 72°C for 30s; completion step of 72°C for 5min. Cycle numbers may be reduced to prevent reaching the plateau phase of PCR amplification if semiquantitative analysis is required.
- Analyse RT-PCR products by agarose gel electrophoresis (see Section III,B).
IV. SELECTION OF A SUITABLE METHOD FOR APOPTOSIS DETECTION
There are many factors to be considered when selecting a suitable method for investigating apoptosis. These factors include the cell type being analysed, the nature of the cell death inducer, the stage of cell death, the information required from the study (e.g., whether information on single cells or on the cell population, as a whole, is required), and the resources available (e.g., where or not access is available to a fluorescence microscope, flow/cytometer, thermocycler, etc.). To assist with selection, Table III summarises the "advantages" and "disadvantages" of the procedures detailed in this article. To form a more extensive understanding of the events occurring within cells, it is advisable, whenever possible, to investigate cell death using more than one technique.
 |
References
Ausubel, M. A., Brent, R., Kinston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A., and Struhl, K. (eds.) (1991). Current Protocols in Molecular Biology, pp. 4.0.1-4.0.9. Greene Publishing Associates and Wiley Interscience, New York.
Barinaga, M. (1998). Death by dozens of cut. Science 280, 32-34. Chomczynski, P., and Sacchi, N. (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156-159.
Clynes, M., Daly, C., NicAmhlaoibh, R., Cronin, D., Elliot, C., O'Connor, R., O'Doherty, T., Connolly, L., Howlett, A., and Scanlon, K. (1998). Recent developments in drug resistance and apoptosis research. Crit. Rev. Oncol. Hematol. 28, 181-205.
Darzynkiewicz, Z., and Traganos, E (1998). Measurement in apoptosis. In "Apoptosis: Advances in Biochemical Engineering Biotechnology" (T. Scheper, ed.), pp. 33-73. Springer, New York.
Kerr, J. E R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239-257.
Kiechle, E L., and Zhang, X. (2002). Apoptosis: Biochemical aspects and clinical implications. Clin. Chim. Acta 326, 27-45.
Mahotka, C., Wenzel, M., Springer, E., Gabbert, H. E., and Gerharz, C. D. (1999). Survivin-AEx3 and survivin-2B: Two novel splice variants of the apoptosis inhibitor survivin with different antiapoptotic properties. Cancer Res. 59, 6097-6102.
Majno, G., and Joris, I. (1995). Apoptosis, oncosis, and necrosis: An overview of cell death. Am. J. Pathol. 146, 3-15.
Nagata, S. (1997). Apoptosis by death factor. Cell 88, 355-365. O'Driscoll, L., Daly, C., Saleh, M., and Clynes, M. (1993). The use of reverse transcriptase-polymerase chain reaction (RT-PCR) to investigate specific gene expression in multi-drug resistant cells. Cytotechnology 12, 289-314.
O'Driscoll, L., Kennedy, S., Mc Dermott, E., Kelehelan, P., and Clynes, M. (1996). Multiple drug resistance-related messenger RNA expression in archival formalin-fixed paraffin-embedded human breast tumour tissue. Eur. J. Cancer 32, 128-133.
O'Driscoll, L., Linehan, R., Kennedy, S. M., Cronin, D, Purcell, R., Glynn, S., McDermott, E. W., Hill, A. D., O'Higgins, N. J., Parkinson, M., and Clynes, M. (2003). Cancer Lett. 201(2), 225-36.
O'Driscoll, L., Crowin, D., Kennedy, S. M., Purcell, R., Linehan, R., Glynn, S., Larkin, A., Scanlon, K., McDermott, E. W., Hill, A. D., O'Higgins, N. J., Parkinson, M., Clynes, M. (2004) Anticancer Res. 24(2A):473-82.
Ramachandra, S., and Studzinski, G. P. (1995). Morphological and biochemical criteria of apoptosis. In "'Cell Growth and Apoptosis" (G. P. Studzinski, eds.), pp. 119-142. IRL Press, Oxford.
Sambrook, J., and Russell, D.W. (2001). "Molecular Cloning: A Laboratory Manual," 3rd Ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Sesti, G. (2002). Apoptosis in the beta cells: Cause or consequence of insulin secretion defect in diabetes? Ann. Med. 34, 444-450. Van Cruchten, S., and Van Den Broeck, W. (2002). Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat. Histol. Embryol. 31, 214-223.
Vila, M., and Przedborski, S. (2003). Targeting programmed cell death in neurodegenerative diseases. Nature Rev. Neurosci. 4, 365-375.
Wilson, J. W., and Potten, C. S. (1999). In "Apoptosis. A Practical Approach" (G. P. Studzinski, ed.), pp. 19-39. IRL Press, Oxford University Press.
Support our developers




