Postnatal Skeletal Stem Cells:
Methods for Isolation and Analysis of Bone Marrow Stromal Cells from Postnatal Murine and Human Marrow
I. INTRODUCTIONSkeletal stem cells are found among the adherent and clonogenic subset of bone marrow stromal cells. It is of utmost importance to realize that the very existence of a skeletal stem cell is established through a complex sequence of ex vivo isolation and expansion, and in vivo transplantation. Through the ex vivo expansion of a single cell-derived strain and the subsequent in vivo transplantation, a complete heterotopic bone/bone marrow organ, containing a hematopoiesis-supporting stroma, must be established in order to prove that the single, originally cloned cell was indeed a stem cell. In addition, stromal cells isolated from the heterotopic organ must be able to transfer the hematopoietic microenvironment and have the potential to establish bone tissue in vivo upon serial transplantation. In vivo transplantation of stroma cells from a variety of species usually does not lead to the formation of cartilage. This is due to the conditions of a relatively high oxygen tension established in open transplantation systems. Hence, the chondrogenic potential of the cell strain under examination must be probed separately using in vitro micromass cultures (Bianco and Robey, 2004).
It is also important to realize that monolayers of stromal cells, established through clonal or nonclonal cultures, are not per se homogeneous populations of "stem" cells, but an uncontrolled mixture of cells and progenitors of highly diverse differentiation potential. This is the natural consequence of the natural asymmetric kinetics of stromal stem cell growth in culture, which inherently leads to a progressive dilution of the original stem cells present in the explanted cells, unless some degree of expansion of the stem cells is allowed, during culture, through the stochastic reversal of asymmetric kinetics to symmetric expansion.
In vitro and in vivo assays are necessarily complementary to one another in the study of the biology and pathology of skeletal stem cells and cannot be sensibly used in isolation. Most in vitro assays of differentiation potential do not necessarily predict the behavior of the same test strain upon in vivo transplantation.
II. MATERIALS AND INSTRUMENTATION
Mice of any strain, including transgenic lines, can be used as a source of bone marrow stromal cells (BMSCs). Guinea pigs (Hartley Davis), used to create irradiated feeder cells, are obtained from Charles River Laboratories. Human bone fragments are collected as surgical waste, and human bone marrow aspirates are obtained from normal volunteers, both under internal review board-approved protocols for the use of human subjects in research. For in vivo transplantation experiments, female Bg Nu/Nu-Xid mice, between 6 weeks and 6 months of age, are from Harlan. Standard tissue culture supplies (tubes, pipettes, dishes, flasks) and solutions [α-MEM, Coon's modified Ham's F-12, Hanks' balanced salt solution (HBSS), trypsin/EDTA, glutamine, penicillin-streptomycin] are not vendor specific. Cell strainers are from Becton-Dickinson (Cat. No. 2350). Lot-selected fetal bovine serum is obtained from a number of vendors (see Section V). Chondroitinase ABC is obtained from Seikagaku America (Cat. No. 100330-1A). Cloning cylinders were obtained from Sigma (Cat. No. C3983). Recombinant human TGFβ1 is obtained from Austral Biologics (Cat. No. GF-230-2). Hydroxyapatite/ tricalcium ceramic, particle size 0.5-1 mm, is obtained from Zimmer by a material transfer agreement, and Gelfoam is from Upjohn (dental packs, size 4, 2 × 2 cm, Cat. No. NDC 0009-0396-04). Mouse fibrinogen and thrombin are from Sigma (Cat. Nos. F-4385 and T- 8397, respectively). Ketamine hydrochloride, xylazine hydrochloride, and acepromazine (Cat. Nos. K2753, X1251, and A6908, respectively) are from Sigma. All other standard chemicals and reagents are from Sigma. Polymerase chain reaction is performed using a commercially available kit from Roche Diagnostics (Cat. No. 1 636 103). Standard equipment for use in dissection (scissors, forceps, blade knives) are sterilized by autoclaving prior to use and can be obtained from any vendor. Cell number enumeration is determined by use of a standard hemocytometer. Cell cultures are viewed by standard inverted and dissecting microscopes, and tissue sections are viewed by standard bright-field microscopes.
III. PROCEDURES
Bone marrow stromal cells can be prepared from bone specimens or bone marrow aspirates from any animal species using a variety of procedures. While there have been a number of modifications whereby single cell suspensions of marrow are subfractionated by density gradient centrifugation, the original assay described by Friedenstein (and described later) relies on the rapid adherence of BMSCs to plastic and avoids loss of these cells during fractionation (Friedenstein, 1980; Friedenstein et al., 1992; Kuznetsov et al., 1997a,b). When working with human cells, BMSCs can be isolated by FACS using the mouse monoclonal antibody Stro-1. However, there is significant contamination by hematopoietic cells such that adherence to tissue culture plastic is necessary to purify them further. It must also be noted that culture conditions must be optimized for each animal species that is used, particularly, in selecting appropriate lots of serum. The procedures presented here focus on the preparation of mouse and human cells in particular due to the fact that they are used most frequently, and to highlight some of the differences between establishing cultures from these two different species.
A. Collection and Preparation Single Cell Suspensions of Bone Marrow
Solutions
1. Nutrient medium: α-MEM
2. Heparinized nutrient medium: α-MEM containing 100 U / ml sodium heparin.
Steps
- For preparation of mouse and guinea pig marrow, animals are euthanized by CO2 inhalation in compliance with institutionally approved protocols for the use of animals in research. Femora, tibiae, and humeri are removed aseptically, and the entire bone marrow content of medullary cavities is flushed with nutrient medium and combined. From human surgical specimens, trabecular bone fragments are scraped with a steel blade into the nutrient medium and washed until the bone became marrow free. In other cases, a 0.5-ml aspirate is collected and mixed with 5ml of ice-cold nutrient medium containing 100U/ml sodium heparin. The cells are centrifuged at 135g for 10min, and the pellet is resuspended in fresh nutrient medium.
- To prepare single cell suspensions (from all animal species), marrow preparations are pipetted up and down several times, passed through needles of decreasing diameter (gauges 16 and 20) to break up aggregates, and subsequently filtered through a cell strainer. Excessive pressure, both positive and negative, should be avoided while passing cell suspensions through the needles. Mononuclear cell concentrations are determined with a hemocytometer.
- Guinea pig marrow suspensions, used as feeder cells in mouse cultures, are γ-irradiated with 6000cGy to prevent the proliferation of adherent guinea pig cells.
B. Determination of Colony-Forming Efficiency (enumeration of CFU-F)
The concentration of CFU-F in bone marrow is usually expressed as the colony-forming efficiency (CFE), or number of BMSC colonies per 1 × 105 marrow nucleated cells in the original marrow cell suspension. In animals under physiological conditions, CFE remains relatively stable; it is, however, somewhat age dependent and can be altered significantly by experimental procedures, such as acute bleeding, irradiation, or curettage (Friedenstein, 1976, 1990). In humans, CFE is also relatively constant; in normal bone marrow not diluted with peripheral blood, as occurs when aspirated, it is between 20 and 70 per 1 × 105 marrow cells (Kuznetsov and Gehron Robey, 1996).
Solutions
1. Serum-containing medium (SM): SM consists of α- MEM, glutamine (2 mM), penicillin (100 U/ml), streptomycin sulfate (100µg/ml), and 20% lot-selected fetal bovine serum.
2. Hanks' balanced salt solution
3. 100% methanol
4. Saturated methyl violet
Steps
- Mouse cells (6-15 × 105 nucleated cells) or human cells (1-6 × 105 nucleated cells) are plated into 25-cm2 plastic culture flasks in 5ml of SM. If significantly abnormal CFE can be expected, as in some human pathologies, numbers of nucleated cells per flask should be adjusted accordingly. In problematic cases, it is recommended that several groups of flasks, containing, for example, 1 × 104, 1 × 105 , and 1 × 106 nucleated cells, are prepared.
- After 2-3hr of adhesion, unattached cells are removed by aspiration, and cultures are washed vigorously three times with SM. No more than several hundred nonadherent cells remain after the washing step.
- Each flask receives 5 ml of SM. For mouse cultures, irradiated guinea pig feeder cells (1.0-1.5 × 107 nucleated cells per flask) are added. If no more than 1 x 105 human cells per 25-cm2 flask are plated, steps 2 and 3 can be omitted.
- Cultivation is performed at 37°C in a humidified atmosphere of 5% CO2 with air. On days 10-14, cultures are washed with HBSS, fixed with methanol, and stained with an aqueous solution of saturated methyl violet.
- Colonies containing 30 or more cells are counted using a dissecting microscope, and colony-forming efficiency (number of colonies per 1 × 105 marrow cells plated) is determined (See Fig. 1A).
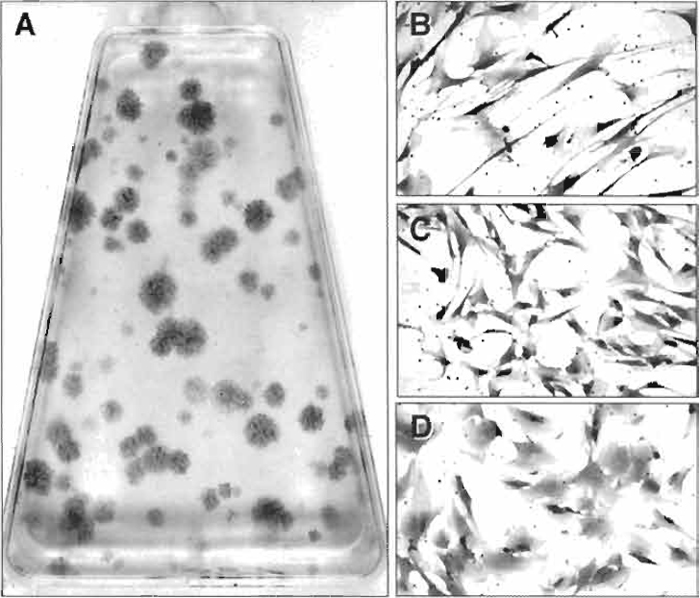 |
| FIGURE 1 (A) When single cell bone marrow suspensions are plated at low density, the colony-forming
unit-fibroblast present in the cell population adheres and forms colonies of bone marrow stromal cells. Their
enumeration at low density is the basis of the colony-forming efficiency assay. Colonies are heterogeneous
and grow at different rates, generating a broad range of colony sizes that are composed of cells with different
morphologies, ranging from elongated, spindle-shaped cells (B), cells with a more compact morphology (C), and very flat and extended cells (D). |
C. Preparation of Multicolony.Derived Strains of BMSCs
Solutions
- Hanks' balanced salt solution
- Chondroitinase ABC: Dissolve powdered chondroitinase ABC in HBSS to achieve a concentration of 20mU/ml, aliquot, and store at-20°C.
- Serum-containing medium: SM is prepared as described earlier.
- Trypsin/EDTA: 0.05% trypsin with 0.53 mM EDTA
- Fetal bovine serum (FBS)
Steps
- Suspensions of mouse cells are prepared as described earlier. Contents of six mouse bones (two each of femora, tibiae, and humeri, approximately 6-8 × 107 nucleated cells) are plated per 75-cm2 flask. Suspensions from human surgical specimens are plated at 5 × 106 to 5 × 107 nucleated cells, and suspensions from aspirates are plated at 5 × 106 to 20 × 107 nucleated cells in 75-cm2 flasks or 150mm 2 dishes containing 30-50 ml of SM.
- Cells are cultured at 37°C in a humidified atmosphere of 5% CO2 with air, and medium is replaced on day 1 for human aspirates and at day 7 for all cultures. Passage generally is performed on days 12 to 14.
- The resulting mouse cultures are passaged by (a) washing twice with HBSS, (b) incubating with chondroitinase ABC for 25-35 min at 37°C, (c) washing with HBSS, (d) treatment with trypsin/EDTA for 25-30rain at room temperature, (e) a second treatment with trypsin/EDTA for 25-30rain at 37°C, and (f) a final wash with SM. Steps b and c are omitted after passages greater than two. Human cultures are washed twice with HBSS and treated with two consecutive applications of trypsin/EDTA for 10 to 15rain each at room temperature, followed by a wash with SM.
- Cold FBS is added to each fraction as collected (final concentration 1%) to inhibit enzymatic activity. Fractions are combined, pipetted vigorously to break up cell aggregates, and centrifuged at 135g for 10rain, and the cell pellet is resuspended in fresh SM. Mouse cells are plated at 2-10 × 106 cells per 75-cm2 flask depending on hematopoietic cell numbers. The next passage is performed when cultures approach confluency. Human cells are plated at 2 × 106 cells per 75-cm2 flask or 150mm dish. Upon reaching approximately 70% confluency, cells are passaged using the same procedure.
D. Establishment of Single Colony-Derived Strains of BMSCs
While multicolony derived strains of BMSCs take on a homogeneous appearance after passaging, and their differentiation potential can be characterized en mass, examination of colonies that form when bone marrow suspensions are plated at low density (as in the CFE assay, Fig. 1A) shows a great deal of heterogeneity in the starting population. There is a marked difference in the growth rate of the colonies, as demonstrated by colonies of different size. Furthermore, colonies are formed by cells with varying morphologies, ranging from a long, spindle shape (Fig. 1B) to a more compact shape (Fig. 1C) and to a very flat and spread morphology (Fig. 1D). Based on this heterogeneity, a number of studies have focused on the characterization of single colony-derived strains, prepared as described later (Kuznetsov et al., 1997b), in order to better understand the hierarchy of BMSCs and their differentiation potential.
Solutions
1. Serum-containing medium: SM is prepared as described earlier.
2. Hanks' balanced salt solution
3. Trypsin/EDTA
Steps
- Mouse cells, 6-15 × 105 nucleated cells, are plated in 150-mm petri dishes in order to prepare single colony-derived strains. From human surgical specimens, 0.007-3.5 × 103 nucleated cells/cm2, and from aspirates, 0.14-14.0 × 103 nucleated cells/cm2, are plated in 150-mm dishes containing 30-50ml of SM. Cells may also be plated by limiting dilution in 96-well microtiter plates.
- After adhesion for 2-3hr, cultures are washed vigorously, and irradiated guinea pig cells are added to mouse cultures as described earlier.
- After 14 to 16 days, cultures are inspected visually, and well-separated colonies of perfectly round shape are identified for cloning. The medium is removed, cultures are washed with HBSS, and individual colonies are surrounded by a cloning cylinder attached to the dish with sterilized high vacuum grease.
- Cells inside the cylinder are treated with two consecutive aliquots of trypsin/EDTA for 5-10min each at room temperature. In both cases, the released cells are transferred to individual wells of 6-well plates containing SM.
- Subsequent passage is performed before cells reach confluence, usually 5 to 10 days later. Each strain is passaged consecutively to a 25-cm2 flask (second passage) and to a 75-cm2 flask (third passage).
E. Cartilage Formation by BMSCs in Micromass (Pellet) Cultures
Cartilage formation by BMSCs is generally performed in vitro using high-density "pellet" cultures, which generate a relatively anaerobic environment that is conducive for chondrogenesis, along with a chondrogenic medium. The following procedure is essentially as described by Johnstone et al. (1998), although it has been suggested that cells grown from day 0 with basic fibroblast growth factor (FGF-2) display more chondrogenic potential (Muraglia et al., 2003).
Solution
1. Chondrogenic medium: Coon's modified Ham's F-12 medium is supplemented with 10-6M bovine insulin, 8 × 10-8M human apo-transferrin, 8 × 10-8M bovine serum albumin, 4 × 10-6M linoleic acid, 10-3M sodium pyruvate, 10ng/ml rhTGFβ1, 10-7M dexamethasone, 2.5 × 10-4M ascorbic acid, or media with similar formulation.
Steps
- Either multicolony-derived or single colonyderived BMSCs (2.5 × 105) are centrifuged at 500g in 15-ml polypropylene conical tubes in 5 ml of chondrogenic medium.
- Cultures are incubated with caps partially unscrewed for 3 weeks at 37°C in 5% CO2, with a medium change at 2- to 3-day intervals. Pellets should not be attached to the tubes.
- At harvest, pellets are washed with phosphatebuffered saline (PBS), fixed in 4% neutral-buffered formaldehyde for 2h, and embedded in paraffin for histological analysis (Figs. 3A and 3B).
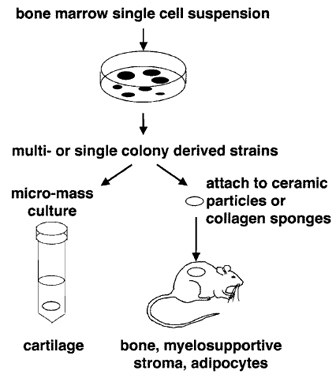 |
| FIGURE 2 BMSC colonies can be passaged together to form multicolony-derived strains or isolated individually to form single colony-derived strains. Both types of cultures can be used to form cartilage in micromass (pellet) cultures in the presence of a chondrogenic medium and to demonstrate the ability to form bone, myelosupportive stroma, and adipocytes by in vivo transplantation in association with hydroxyapatite/tricalcium phosphate particles or collagen sponges. |
In vivo transplantation of BMSCs has become the gold standard by which to measure their multipotentiality (see Fig. 2). In conjunction with hydroxyapatite/ tricalcium phosphate (HA/TCP) or collagen sponges as described by Krebsbach et al. (1997), BMSCs have the ability to form bone, myelosupportive stroma, and adipocytes, thereby recreating an ectopic bone/marrow organ (ossicle) when transplanted subcutaneously into immunocompromised mice. It should be noted that while human BMSCs are not as sensitive to culture conditions as murine BMSCs, they are more sensitive to the substrates used for in vivo transplantation. To date, HA/TCP particles appear to provide the best substrate for ossicle formation by human BMSCs (Figs. 3C and 3D).
Solutions
- Serum-containing medium: SM is prepared as described earlier.
- Mouse fibrinogen: Mouse fibrinogen is reconstituted in sterile PBS at 3.2mg/ml, aliquoted, and kept at -80°C.
- Mouse thrombin: Mouse thrombin is reconstituted in sterile 2% CaCl2 at 25U/ml, aliquoted, and kept at -80°C. (Because the salt is CaCl2.2H20, in order to prepare 2% solution, 2.65 g of the salt should be diluted in 100 ml of water.)
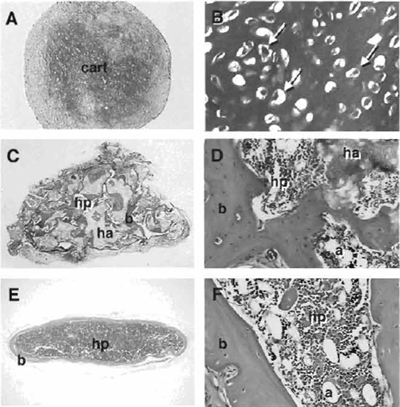 |
| FIGURE 3 In micromass (pellet) cultures in chondrogenic medium, human BMSCs form a dense extracellular matrix that is metachromatic when stained with toluidine blue (A) and features chondrocyte-like cells embedded in this matrix (arrows in B). When transplanted in conjunction with hydroxyapatite/tricalcium phosphate particles (ha), human BMSCs form bone (b) that is actively deposited on the surface of the particles (C) and, with time, establish a fully functional hematopoietic marrow (hp), including adipocytes (a) as shown (D). Murine BMSCs are also able to form a cortex of bone (E) that surrounds a complete hematopoietic marrow with adipocytes when transplanted in collagen sponges (F). |
Steps
- Sterilization of HA/TCP particles is achieved by placing the particles into a glass bottle sealed with foil and heating at 220°C overnight (minimum 8h). The particles are then aliquoted (40-mg/sterile roundbottomed centrifuge tube) in a hood using a sterile balance, sterile weighing spatula, and sterile filter paper.
- Passaged BMSCs (see earlier discussion) are counted, pelleted at 135g for 10min, and resuspended in SM so that the volume in milliliters is equal to the number of transplants to be prepared.
- HA/TCP powder is washed twice with SM, and the medium is discarded.
- BMSCs designated for an individual transplant (1-2 × 106 cells in 1 ml of SM) are transferred into a tube with HA/TCP powder. The powder is mixed with the cell suspension and is incubated at 37°C for 70-100 min with slow rotation (25 rpm).
- Particles with adherent BMSCs are collected by a brief centrifugation (135g for l min), and the supernatant is removed carefully.
- Mouse fibrinogen (15 µl) is added to the cell/particle mixture, and the components are mixed gently. Mouse thrombin (15µl) is added to the cell/particle mixture, and the components are mixed gently.
- The tubes are left for a few minutes at room temperature. After a clot is formed, the cap is closed tightly to prevent the transplant from drying. The resulting fibrin clot with HA/TCP powder and BMSCs has the consistency of a gel; it can be easily taken out of the tube with a spatula and placed into a recipient animal as described later.
G. Preparation of BMSC and Collagen Sponge Constructs
Murine BMSCs generate complete ossicles in conjunction with HA/TCP and also when transplanted in collagen sponges (Gelfoam) (Figs. 3E and 3F), whereas human BMSCs only generate limited amounts of bone in the latter substrate.
Solution
1. Serum-containing medium: SM is prepared as described earlier.
Steps
- Sterile Gelfoam sponges are cut into cubes of the desired size or into any other shape, placed into SM, and squeezed with forceps so that they regain their full size but with no air bubbles left inside.
- BMSCs designated for individual transplants are transferred (1-2 × 106 cells/1 ml of SM) into an individual Eppendorf tube, pelleted at 135g for 10min, and most of the supernatant is discarded. The volume left should be smaller than the volume of the sponge (usually about 50 µl). The pellet is resuspended in this small volume.
- Individual sponges are blotted between two sheets of sterile filter paper and are placed immediately into freshly resuspended cells in an Eppendorf tube where it expands, capturing the cell suspension. The tube is sealed tightly to prevent drying.
- The sponges are incubated at 37°C for 90min.
- Sponges with cells are transplanted as described
later.
H. In Vivo Transplantation of BMSC Constructs
Solutions
1. Anesthesia: Combine 225µl ketamine, 69~tl of xylazine, 75µl of acepromazine, and 231 µl of H2O (total volume= 600 µl). Use 100 µl/mouse (25 g). If mice are smaller, less anesthesia should be used.
2. Betadine
3. 70% ethanol
Steps
- Anesthetize the mouse and clean the skin with betadine and 70% ethanol. A single 3-cm-longitudinal incision is made in the skin along the dorsal surface.
- The tip of the dissecting scissors is used to make a pocket for the transplant by inserting the scissor subcutaneously and then opening the scissors approximately I cm. A sterile spatula is used to insert HA/TCP transplants, and forceps are used to insert collagen sponge transplants. The procedure is repeated twice on each side of the incision for a total of four pockets. The incision is closed with several autoclips. Autoclips are not removed due to the fact that it causes extensive bleeding in immunocompromised mice.
- Transplants are harvested at various time points, fixed with 4% neutral-buffered formaldehyde overnight, decalcified, and embedded in paraffin for standard histological analyses.
- Because both HA/TCP and collagen sponge transplants are open systems, there is no barrier to prevent host cells from invading transplants. Determination of cells of donor origin in transplants generated by murine cells requires that the donor bear a marker (e.g., lacZ, green fluorescent protein, or a transgene) (Krebsbach et al., 1997). When human cells are used, antibodies that recognize human proteins but not mouse analogs are commonly used (Krebsbach et al., 1997). In situ hybridization using human-specific Alu sequences as the probe has been particularly useful in characterizing transplants generated by human cells (Kuznetsov et al., 1997).
IV. COMMENTS
The methods of ex vivo expansion presented here are expressly for maintaining BMSCs in an uncommitted state. Subsequently, it is possible to demonstrate that these cells, either multicolony-derived or some single colony-derived strains, have the ability to generate multiple phenotypes. Their chondrogenic potential can be demonstrated in vitro by the establishment of high-density pellet cultures with a chondrogenic medium, and their ability to form bone, myelosupportive stroma, and adipocytes is best assessed by transplantation in vivo. These results demonstrate that a true postnatal stem cell exists within the population of BMSCs, with well-known implications in the emerging field of regenerative medicine. Furthermore, in vivo transplantation of BMSCs bearing gene defects, either occurring naturally or created by molecular engineering, provides the opportunity to study the specific role of a gene in the process of establishing a bone/marrow organ (Bianco and Robey, 1999).
While the differentiation capacity of BMSCs is best evaluated by in vivo transplantation, there are many methods for inducing an osteogenic phenotype in vitro. However, it must be noted that these methods result in the formation of a tissue that does not have the structural organization of bone that is formed in vivo; in many cases, mineralization is due to dystrophic calcification as opposed to true bone formation. Adipogenesis can also be induced in vitro by a variety of different culture modifications (reviewed in Bianco et al., 1999), but again, the adipocytes that are formed tend to be multivacuolar (immature), whereas mature adipocytes in marrow are univacuolar. Nonetheless, cultures of BMSCs provide the opportunity to study the effects of extrinsic signals that cause these cells to shift from one phenotype to another and to analyze the resultant changes in the pattern of gene expression.
V. PITFALLS
- The specific lot of fetal bovine serum used is critical not only for the determination of CFE, but also for the ex vivo expansion of BMSCs. It is not well recognized that fetal bovine serum must be tested extensively to select lots that are suitable for one animal species or another.
- In murine BMSC cultures (and from other rodents), macrophages represent a major contaminant in the adherent population. In low-density cultures, clusters of macrophages are often mistaken by the untrained eye for a colony established by a CFU-E In cultures established by high-density plating, ~11% of the adherent population are macrophages (as identified by {x-naphthyl acetate esterase activity) at the second passage, and further passage or cell sorting is needed to eliminate them (Krebsbach et al., 1997). The contamination by macrophages in murine cultures not only has an impact on determination of CFE, but also on the interpretation of a variety of in vitro studies intending to determine the direct effect of factors on BMSCs and in vivo studies aimed at identifying where BMSCs engraft and what cell types they form after systemic injection.
- In establishing BMSCs from bone marrow aspirates, the volume should not exceed 0.5cc from any one site, or without repositioning the needle. Drawing larger volumes results in contamination of the marrow by peripheral blood, which influences the determination of CFE. Furthermore, peripheral blood has a negative influence on the growth of BMSCs.
References
Bianco, P., Riminucci, M., Kuznetsov, S., and Robey, E G. (1999). Crit. Rev. Eukaryot. Gene Expr. 9, 159-173.
Bianco, P., and Robey, P. (1999). J. Bone. Miner. Res. 14, 336-341.
Bianco, P., and Robey, P. G. (2004). Skeletal stem cells. In "Handbook of Adult and Fetal Stem Cells" (R. P. Lanza, ed.). Academic Press, Sam Diegs.
Friedenstein, A. J. (1976). Int. Rev. Cytol. 47, 327-359.
Friedenstein, A. J. (1980). Hamatol. Bluttransfus. 25, 19-29.
Friedenstein, A. J. (1990). Osteogenic stem cells in bone marrow. In "Bone and Mineral Research" (J. N. M. Heersche, J. A. Kanis, eds.), pp. 243-72. Elsevier, New York.
Friedenstein, A. J., Latzinik, N. V., Gorskaya Yu, E, Luria, E. A., and Moskvina, I. L. (1992). Bone Miner. 18, 199-213.
Johnstone, B., Hering, T. M., Caplan, A. I., Goldberg, V. M., and Yoo, J. U. (1998). Exp. Cell Res. 238, 265-272.
Krebsbach, P. H., Kuznetsov, S. A., SatomuraK., Emmons, R. V., Rowe, D. W., and Robey, P. G. (1997). Transplantation 63, 1059-1069.
Kuznetsov, S., and Gehron Robey, P. (1996). Calcif. Tissue Int. 59, 265-270.
Kuznetsov, S. A., Friedenstein, A. J., and Robey, P. G. (1997a). Br. J. Haematol. 97, 561-570.
Kuznetsov, S. A., Krebsbach, P. H., Satomura, K., Kerr, J., Riminucci, M., et al. (1997b). J. Bone Miner. Res. 12, 1335-1347.
Muraglia, A., Corsi, A., Riminucci, M., Mastrogiacomo, M., Cancedda, R., et al. (2003). J. Cell Sci. 116, 2949-2955.
Support our developers




