Preparation and Immortalization of Primary Murine Cells
I. INTRODUCTIONThe generation of immortal cell lines from genetically defined mice has proven useful in the understanding of molecular pathways in mammalian biology. This article describes the preparation and immortalization of murine embryo fibroblasts (MEFs), the workhorse cell type of genetically defined mice. These cells are used most frequently because of their ease of preparation, relative homogeneity, and high frequency of immortalization with serial passage in culture. These techniques can be used, with modification, to immortalize rat embryo fibroblasts as well. Furthermore, variations of these techniques can be applied to other murine cell types (e.g., astrocytes or keratinocytes) for the study of cell-type specific pathways (see Section V).
The genetics of immortalization in murine cells are now largely understood [Fig. 1, reviewed in Sharpless and DePinho (1999)]. As opposed to human cells, the spontaneous immortalization of cultured rodent cells occurs with high frequency resulting from a stochastic genetic event. This difference in frequency is attributable to the additional requirement in human cells for telomerase expression as detailed in the previous article. In murine cells, the act of cell culture, with a few exceptions, potently induces both antiproliferative products (p16INK4a and p19ARF) of the Ink4a/Arf locus, which regulate the Rb and p53 pathways, respectively (Fig. 1). Immortalization of murine cells requires circumventing at least p19ARF-p53 or, in some cell types, both of the pathways regulated by the products of the Ink4a/Arf locus (for examples, see Bachoo et al., 2002; Kamijo et al., 1997; Randle et al., 2001). This can be done in one of three ways: spontaneous, stochastic genetic deletion through serial culture; the use of immortalizing oncogenes; or the use of genetically defined mice. This article deals predominantly with the former method, the latter two are described in Section V.
![FIGURE 1 The genetics of senescence. In murine cells, the act of culture [("culture shock" (Sherr and DePinho, 2000)] induces both products of the Ink4a/Arf locus, p16INK4a inhibits cyclin-dependent kinases 4 and 6, leading to Rb hyphophorylation and growth arrest. p19ARF stabilizes p53 by inhibiting its mdm2-mediated degradation; p53 also induces growth arrest. In MEFs, the p19ARF-p53 axis dominates the growth phenotype (Kamijo <em>et al.</em>, 1997; Sharpless <em>et al.</em>, 2001), although both p16INK4a and p19ARF contribute to senescence in other cell types (Bachoo <em>et al.</em>, 2002; Randle <em>et al.</em>, 2001).](images/v1_pa_s05_c28_f01.jpg) |
| FIGURE 1 The genetics of senescence. In murine cells, the act of
culture [("culture shock" (Sherr and DePinho, 2000)] induces both
products of the Ink4a/Arf locus, p16INK4a inhibits cyclin-dependent
kinases 4 and 6, leading to Rb hyphophorylation and growth arrest.
p19ARF stabilizes p53 by inhibiting its mdm2-mediated degradation; p53 also induces growth arrest. In MEFs, the p19ARF-p53 axis dominates the growth phenotype (Kamijo et al., 1997; Sharpless et al., 2001), although both p16INK4a and p19ARF contribute to senescence in other cell types (Bachoo et al., 2002; Randle et al., 2001). |
II. MATERIALS
-Sterile 10-cm dishes (Falcon Cat. No. 353003)
-Sterile 6-cm dishes (Falcon Cat. No. 353002)
-Sterile phosphate-buffered saline (PBS, GIBCO Cat. No. 14190-144)
-100 and 70% ethanol
-0.25% trypsin-EDTA (GIBCO Cat. No. 25200-056).
-DMEM (with glucose and L-glutamine, GIBCO Cat. No. 11995-065)
-Heat-inactivated fetal calf serum (FCS, Sigma Cat. No. F-2442; to heat inactivate, thaw and then place in water bath at 56°C for 30min)
-100× penicillin/streptomycin (P/S 10,000 units/ml, GIBCO Cat. No. 15140-122)
-Optional: β-mercapoethanol (β-ME, Sigma Cat. No. M-7522)
-Liquid nitrogen-safe cryotybes (Corning Cat. No. 2028)
-Dimethyl Sulfoxide (DMSO, Mallinkrodt AR Cat. No. 4948)
III. INSTRUMENTATION (ALL STERILE)
- 6 ¾in. Mayo scissors (Fisher Cat. No. 13-804-8)
- Large forceps (Fisher Cat. No. 13-812-36)
- Curved fine scissors (Fisher Cat. No. 08-951-10)
- Two curved small forceps (Fisher Cat. No. 08-953D)
- Two 5 ½ in. Kelly clamps (Fisher Cat. No. 08-907)
- 9-in. cotton-plugged Pasteur pipettes (Fisher Cat. No. 13-678-8B).
- Razor blades (place in 100% ethanol in 15-m dish prior to use)
IV. PROCEDURES
A. Murine Embryo Fibroblast Production
Day 1
- The embryo dissection need not be performed in the hood but rather can be done on a clean bench covered with a diaper. Use sterile reagents (e.g., autoclave or flame dissecting equipment).
- Sacrifice the pregnant mother at approximately noon on postcoital day 13.5 (see Section VI) through CO2 inhalation. If the embryos are of potentially different genotypes (e.g., the progeny of a heterozygous intercross), regenotype the mother at this stage.
- Place the mother on a dissection stand and clean by spraying generously with 70% ethanol.
- Using Mayo scissors and large forceps, carefully open the abdomen, locate the embryos, and remove the bicornuate uterus with embryos. The embryos will appear as ~l-cm "beads on a string," in general there will be 6-12 embryos in a normal litter. If a mating produces an embryo with a genotype associated with developmental lethality, it is often possible to see partially resorbed embryos at this time.
- Place uterus plus embryos in sterile PBS in 10-cm dish. Using fine forceps and/or scissors, gently dissect each balloon-like embryo with fetal membranes from the thicker uterus and place individually into a 6-cm dish with sterile PBS. If there are a large (>12) number of embryos, place the 6-cm dishes on ice. If the fetal membranes rupture during dissection, separate the embryos from the placenta and membranes and put in a 6-cm dish in PBS. Some investigators use a dissecting microscope for this step, although we do not find this necessary.
- Perform the rest of the dissection in a tissue culture hood.
- Dissect away placenta and fetal membranes. Of note, the placenta is partly of maternal origin and can therefore contaminate genotyping if the mother's genotype differs from those of the embryos. To remove placenta, use a pair of fine forceps. Grasp umbilical vessels from the placenta to the embryo with one set of forceps and grasp the placenta with the other and pull in opposite directions. Pulling the placenta directly can cause the abdominal viscera to herniate and detach. The fetal membranes may still be wrapped around the embryo after placental removal, but these can be gently pulled away from the embryo after placental detachment. The fetal membranes are of embryonic origin and can be used for genotyping, although we prefer to use nonadherent cells obtained on day 2 (see later).
- Place the entire embryo without membranes or placenta in a 10-cm dish with 1 ml 0.25% trypsin-EDTA (this is preferable to the 0.05% trypsin-EDTA commonly used to passage cells).
- Hold a razor blade in each of two Kelly clamps and flame after dipping in 100% ethanol. Briefly cool razor blades in sterile PBS in a 10-cm dish and then use to mince the embryos. The pieces of tissue need not be too fine; chopping approximately 30 times per embryo is generally adequate. Change razor blades between embryos if the embryos are of differing genotypes.
- Allow minced tissue to sit in trypsin-EDTA for 10min at 37°C, 5% CO2.
- Add 2ml of growth media (DMEM + 10% FCS + pen/strep with 50µM β-ME; the β-ME is optional) and disaggregate by repeated pipetting: 10 times with a 5-ml pipette and 10 times with a plugged Pasteur pipette. After disaggregation, there should be no large (<1mm in largest dimension) chunks of tissue remaining.
- Add 8 ml of growth media and swirl plates for even seeding. We grow cells at this stage in an incubator dedicated for primary cells in the rare event of bacterial contamination.
Day 2
- The 10-cm dish should be 100% confluent after attachment of MEFs overnight. If subconfluent, this generally indicates poor disaggregation of the embryo, improper media, or bacterial contamination (see Section VI).
- Gently remove the media and save nonadherent cells for genotyping (see later). Take care as the cells are poorly adherent at this stage and large chunks of the monolayer can detach with poor handling.
- Add 10 ml of fresh growth media.
- If genotyping is necessary (i.e., the embryos are of different genetic composition), this can be done on DNA prepared from nonadherent cells collected on day 2. This is done by pelleting these cells in 15-cm conical tubes (1000rpm × 5 min), detergent lysis, and precipitation of DNA. The exact method used depends on the manner of genotyping (e.g., polymerase chain reaction vs Southern blot). One can easily obtain >100 µg of good-quality DNA from these pellets if needed. We prepare using a commercially available DNA extraction kit (Promega #Al125).
Day 3
- The culture should be superconfluent on day 3 and ready to freeze down or passage for further experiments. If cells are less than 100% confluent by day 3, freeze down on day 4.
- To freeze, wash cells once with sterile PBS and add 1 ml of 0.25% trypsin-EDTA. Cells will detach in 5-10min and then add to a 15-ml conical tube with 10 ml of growth media.
- Spin cells at 1000rpm for 5 min.
- Resuspend cell pellet in 3 ml of ice-cold freezing media (90% FCS + 10% DMSO) and put in three welllabeled, chilled cryovials. We label with date prepared, the line number, and the passage number upon thawing. The lines are numbered as the mother's number first, followed by the embryo number (e.g., 115-7 would be the seventh embryo of mother 115). The cells will be passage 2 upon thawing if not passaged further before freezing.
- Keep cells on ice for 10-15 minutes and then put in a -80°C freezer in a sealed styrofoam block overnight. Alternatively, the cells can be placed in a cryo-freezing container (Nalgene Cat. No. 5100-001) and then placed in a -80°C freezer overnight.
- The day after freezing, cells should be moved to a liquid nitrogen tank for long-term storage.
B. Thawing MEFs
- Keep cells on dry ice until absolutely ready to thaw.
- Place the vial of cells in a 37°C water bath and watch carefully. As soon as the cells begin to thaw, pour the entire pellet into a 15-ml conical tube.
- Add 10 ml of growth media dropwise over about 30s with gentle mixing while adding. Mix well after all 10ml of media is added.
- Spin cells down (1000rpm for 5min), resuspend in 3 ml media, and plate in a 10-cm dish. One vial of frozen cells can go into one 10-cm dish; add another 7 ml media.
- The cells should be 90-100% confluent the day after thawing. If confluence is less with large numbers of floating (dead) cells noted, this suggests improper freezing, improper storage (e.g., freezer malfunction), or improper thawing (see Section VI).
C. Immortalizing MEFs
This can be done using a rigorously defined passaging protocol (e.g., 3T3 or 3T9 assay) or, more simply, by serial replating of cells. The advantage of the passaging protocol is that one learns about the growth kinetics and immortalization frequency of the cell (see Section V). The immortalization of wild-type MEFs requires a stochastic genetic event: generally p53 loss or, less frequently, loss of p19ARF (Kamijo et al., 1997). Other immortalizing events, such as mdm2 amplification (Olson et al., 1993), have also been noted, but these are considerably rarer. In our experience, this genetic event is most likely to be loss of p53 (~60-70% of immortalized lines) o r p19ARF (~20-30%) regardless of growth conditions (passage at high or low density). Therefore, serial passaging without counting cells at every passage is an appropriate way to derive immortalized cells. In fact, the "3T3 cell" has become a nondescript term referring to immortal murine embryo fibroblasts obtained through serial passaging, but few laboratories except those interested in growth and senescence do rigorous 3T3 assays to obtain them.
D. Immortalization by Serial Passage
- Split MEFs 1:3 twice per week during the rapid growth phase of the culture (the first 6-10 passages).
- As cells enter senescence, split 1:2 or merely replate (aim to keep cells near 100% confluency on the day of passaging). If the cultures are seeded too sparsely, this will decrease the immortalization frequency.
- Between passages 10 and 20 (4 to 9 weeks in culture), immortalized lines will begin to overgrow the culture. These cells are smaller and spindle shaped, initially appearing as small nests of cells obvious on the day of passaging. These cells can be subcloned, but are used more frequently as pooled immortalized cells.
E. 3T9 Assay
This assay was originally described by Todaro and Green (1963). The "3" refers to passaging every 3 days, and the "9" refers to 9 x 105 cells plated per passage. 3T3 (3 x 105 cells) and 3T12 (1.2 x 10 6 cells) assays can also be done to study growth and immortalization at varying densities. In our experience, immortalization frequency is highest for the 3T3 assay (and therefore this assay has lent its name to all immortalized lines of murine fibroblasts), whereas 3T9 is more useful for measuring immortalization frequency (Sharpless et al., 2001). As stated earlier, serial replating of murine fibroblasts at almost any density will eventually yield immortalized lines as long as the cells are not seeded too sparsely.
- For the first passage, wash cells growing in a 10-cm dish (usually passage 2 at this stage) with PBS, trypsinize (0.05% trypsin-EDTA is adequate for this purpose), dilute into 10ml of growth media, and count.
- Spin down and resuspend cells at a concentration of 9 × 105 cells/ml. Seed 1 ml of cells (9 × 105 cells) into a 6-cm dish and add 2.5 ml of media. Swirl the cells to ensure uniform seeding. Label the dish with the MEF line number and the passage number.
- For subsequent passages, trypsinize and recount cells every 3 days in a manner identical to steps 1 and 2. Record the number of cells counted before each passage. Growth curves and immortalization frequencies can be determined from analysis of these data as described in Section V.
- Immortalized lines will emerge between passages 10 and 20; virtually all lines proliferating after 20 passages will be immortal. If necessary, these lines can be subcloned by limiting dilution, but this is not generally done. The majority of immortalized lines will have lost either p53 or p19ARF function (Kamijo et al., 1997), although various other genetic events can increase or decrease the frequency of immortalization (Frank et al., 2000; Jacobs et al., 1999; Kamijo et al., 1999; Sharpless et al., 2001).
V. COMMENTS
A. Immortalization through the Use of Oncogenes or Genetic Background
A problem with immortalization by serial passage is that the nature of the immortalizing genetic event is not known, although predominantly p53 or p19ARF is inactivated as MEFs escape senescence. As the behavior of p19ARF-deficient cells may be quite different from p53-deficient cells, however, the stochastic inactivation of these pathways may produce significant line-to-line variability and therefore be undesirable. For example, p53 null MEFs are more aneuploid and more resistant to DNA damage than p19ARF null MEFs (Kamijo et al., 1997; Pomerantz et al., 1998; Serrano et al., 1996; Stott et al., 1998). To assure that all lines will evade senescence via a similar mechanism, cells can either be immortalized through the use of an immortalizing oncogene or by using cells derived from animals of a genetic background that resists senescence.
Classically, the most commonly used oncoprotein to immortalize cells is the SV40 large T antigen (Colby and Shenk, 1982; Jat and Sharp, 1986; Todaro and Green, 1966). This molecule inactivates the p53 and Rb pathway, and the majority of murine cell types can be immortalized by transfection or retroviral transduction of TAg. A disadvantage of TAg, however, is that cells expressing this molecule are unstable and are prone to clonal in vitro evolution. Furthermore, as Rb pocket proteins are required for the differentiation of many cells types (Dannenberg et al., 2000), TAg generally impairs or precludes the study of differentiation. Alternatively, MEFs and several other murine cell types can be immortalized with a dominant-negative form of p53 [e.g., p53-DD (Bowman et al., 1996)], which preserves the Rb pathway, although these cells are still more prone to aneuploidy than p19ARF-deficient lines.
The most elegant method of obtaining immortal cell lines is by deriving cells from animals resistant to senescence. The most commonly used strains for this purpose are Ink4a/Arf-deficient (Serrano et al., 1996), p19ARF-deficient (Kamijo et al., 1997), or p53-deficient (Donehower et al., 1992) mice. These strains are widely available and can be obtained from the mouse models of the human cancer consortium (MMHCC http: / / web. ncifcrf, gov / researchresources / mmhcc/). For studies of genetically defined animals, the genetic background of interest can be crossed two generations to mice of these backgrounds and then MEFs (or other cell types) derived as described earlier, which will be immortal in most cases if derived from p53- or p19ARF-deficient embryos. This method can be employed to derive immortal cells from difficult genetic backgrounds that undergo premature senescence in culture (Frank et al., 2000; Jacobs et al., 1999). This strategy can also be employed to obtain cells of nonfibroblast lineages. For example, immortal melanocytes (Chin et al., 1997), skin keratinocytes (unpublished observations), glia (Bachoo et al., 2002), lymphocytes (unpublished observations; Randle et al., 2001), and macrophages (Randle et al., 2001) have been derived successfully from p19ARF- or Ink4a/Arf-deficient mice using standard culture methods for these cell types. To immortalize with high efficiency, cells must be homozygous null for p53, p19ARF, or Ink4a/Arf; therefore, the principal disadvantage of this approach is the extra time needed to backcross to these defined genetic backgrounds.
B. Data Analysis of 3T9 or 3T3 Assay
These data can be plotted as cell number per passage or population doublings (PDs) per passage (Fig. 2). PDs for any given passage = log2(Nf/No) = 1.44* In(Nf/No), where Nf = number of cells counted at the end of the passage and No= number of cells seeded at the beginning of the passage (i.e., 9 × 105 for a 3T9 assay). For the purpose of immortalization frequency, "senescence" occurs if less than 9 × 105 cells are recovered for two consecutive passages. The immortalization frequency = 1-number of senescent lines/total number of lines analyzed. Measured in this way, the immortalization frequency of wild-type MEFs can vary significantly depending on culture conditions, method of embryo preparation, and so on and therefore is only meaningful when compared to proper littermate control embryos analyzed concurrently.
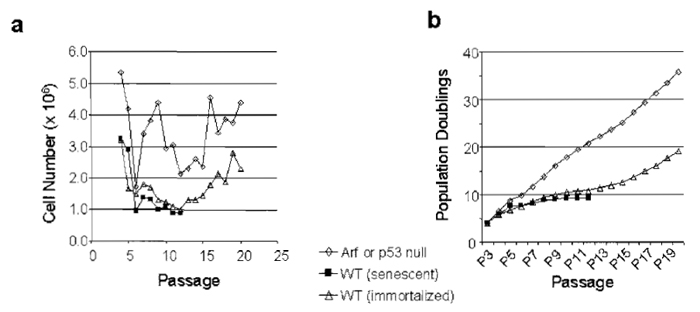 |
| FIGURE 2 The 3T9 assay can be used to quantify both growth and immortalization frequency. Data from a 3T9 (or 3T3) assay can be graphed in either of two ways: (a) as cell number per passage or (b) as population doublings (PDs) per passage (PDs defined in text). The same data are graphed by either method showing a senescent line, an immortalized line, or a line lacking Ink4a/Arf. The p19ARF-dependent slow growth period seen in wild-type MEFs between passages 5-15 is called "senescence," although in actuality it represents a culture-induced phenomena. |
VI. PITFALLS
- Embryo dates. By convention, the day a coital plug is detected is day 0.5 for timed matings, and embryos should be made at midday 13 days later. While it is difficult to be sure of correct plugging dates at the time of dissection, in general day 13.5 embryos have paddle-like front paws, whereas day 14.5 embryos have more fully formed individual digits. Embryos that are too large and well developed or too small suggest an incorrect date of plugging. Cells of different embryonic ages do differ in several in vitro growth properties; therefore, littermate controls are always preferable in MEF experiments. In our experience, MEFs from embryos older than 13.5 grow less well and immortalize less frequently than 13.5 embryos.
- MEFs are not confluent the day after dissection. In general, cells from a single embryo should cover a 10-cm plate fully the morning after plating. Poor coverage of the dish can result from inadequate embryo disaggregation, tissue mincing with razor blades that were not cooled properly after flaming, improper media, or bacterial contamination. In particular, one should be vigilant for bacterial contamination as this can be difficult to note given that there is copious debris in the culture 1 day after plating.
- MEFs are not confluent the day after thawing. This results most often from freezer or liquid nitrogen tank malfunction, but can also be due to improper technique when the cells were frozen, malfunction of the cryo-freezing container, or improper thawing. DMSO is toxic to these primary cells so it is important to resuspend the frozen cell pellet well in growth media, spin the cells down, and then respsund in fresh growth media prior to plating. Thawed vials of cells should not be left in freezing media for a significant period of time prior to replating.
- Cells grow poorly and/or fail to immortalize. This can result from poor growth media (e.g., the fetal calf serum is too old), the use of embryos significantly later than E13.5, or occult pathogen contamination. Embryos of certain genetic backgrounds grow poorly in culture ("premature senescence"), which can sometimes be obviated by backcrossing to Ink4a/Arf-, p19ARF-, or p53-deficient animals (Frank et al., 2000; Jacobs et al., 1999; Kamijo et al., 1999).
References
Bachoo, R. M., Maher, E. A., Ligon, K. L., Sharpless, N. E., Chan, S. S., You, M. J., Tang, Y., DeFrances, J., Stover, E., Weissleder, R., et al. (2002). Epidermal growth factor receptor and Ink4a/Arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1, 269-277.
Bowman, T., Symonds, H., Gu, L., Yin, C., Oren, M., and Van Dyke, T. (1996). Tissue-specific inactivation of p53 tumor suppression in the mouse. Genes Dev. 10, 826-835.
Chin, L., Pomerantz, J., Polsky, D., Jacobson, M., Cohen, C., Cordon- Cardo, C., Horner, J. W., II, and DePinho, R. A. (1997). Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 11, 2822-2834.
Colby, W. W., and Shenk, T. (1982). Fragments of the simian virus 40 transforming gene facilitate transformation of rat embryo cells. Proc. Natl. Acad. Sci. USA 79, 5189-5193.
Dannenberg, J. H., van Rossum, A., Schuijff, L., and te Riele, H. (2000). Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth- restricting conditions. Genes Dev. 14, 3051- 3064.
Donehower, L. A., Harvey, M., Slagle, B. L., McArthur, M. J., Montgomery, C. A., Jr., Butel, J. S., and Bradley, A. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215-221.
Frank, K. M., Sharpless, N. E., Gao, Y., Sekiguchi, J. M., Ferguson, D. O., Zhu, C., Manis, J. P., Horner, J., DePinho, R. A., and Alt, E W. (2000). DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell 5, 993-1002.
Jacobs, J. J., Kieboom, K., Marino, S., DePinho, R. A., and van Lohuizen, M. (1999). The oncogene and Polycomb-group genebmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164-168.
Jat, P. S., and Sharp, P. A. (1986). Large T antigens of simian virus 40 and polyomavirus efficiently establish primary fibroblasts. J. Virol. 59, 746-750.
Kamijo, T., van de Kamp, E., Chong, M. J., Zindy, E, Diehl, J. A., Sherr, C. J., and McKinnon, P. J. (1999). Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res. 59, 2464-2469.
Kamijo, T., Zindy, E, Roussel, M. E, Quelle, D. E., Downing, J. R., Ashmun, R. A., Grosveld, G., and Sherr, C. J. (1997). Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARE Cell 91, 649-659.
Olson, D. C., Marechal, V., Momand, J., Chen, J., Romocki, C., and Levine, A. J. (1993). Identification and characterization of multiple mdm-2 proteins and mdm-2-p53 protein complexes. Oncogene 8, 2353-2360.
Pomerantz, J., Schreiber-Agus, N., Liegeois, N. J., Silverman, A., Alland, L., Chin, L., Potes, J., Chen, K., Orlow, I., Lee, H. W., et al. (1998). The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell 92, 713-723.
Randle, D. H., Zindy, E, Sherr, C. J., and Roussel, M. E (2001). Differential effects of p19(Arf) and p16(Ink4a) loss on senescence of murine bone marrow-derived preB cells and macrophages. Proc. Natl. Acad. Sci. USA 98, 9654-9659.
Serrano, M., Lee, H., Chin, L., Cordon-Cardo, C., Beach, D., and DePinho, R. A. (1996). Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27-37.
Sharpless, N. E., Bardeesy, N., Lee, K. H., Carrasco, D., Castrillon, D. H., Aguirre, A. J., Wu, E. A., Horner, J. W., and DePinho, R. A. (2001). Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413, 86-91.
Sharpless, N. E., and DePinho, R. A. (1999). The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 9, 22-30.
Sherr, C. J., and DePinho, R. A. (2000). Cellular senescence: Mitotic clock or culture shock? Cell 102, 407-410.
Stott, E J., Bates, S., James, M. C., McConnell, B. B., Starborg, M., Brookes, S., Palmero, I., Ryan, K., Hara, E., Vousden, K. H., and Peters, G. (1998). The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 17, 5001-5014.
Todaro, G. J., and Green, H. (1963). Quantitative Studies of mouse embyo cells in cutture and their development into established linos. J. Cell Biol. 17, 299-313.
Todaro, G. J., and Green, H. (1966). High frequency of SV40 transformation of mouse cell line 3T3. Virology 28, 756-759.
Support our developers




