Purification of Intermediate-Containing Spliceosomes
I. INTRODUCTIONThe spliceosome is the dynamic macromolecular machine responsible for removing intervening sequences (introns) from pre-mRNA transcripts. In vitro studies have been used to define a pathway of a spliceosome assembly consisting of several intermediate subcomplexes (Moore et al., 1993). Isolating any one of these splicing complexes for subsequent characterization, and structural analysis requires a means to accumulate that specific complex (Jurica and Moore, 2002). A number of methods for purifying complexes along the spliceosome assembly pathway have been described (Das et al., 2000; Hartmuth et al., 2002; Jurica et al., 2002; Makarov et al., 2002; Zhou et al., 2002). These protocols use similar approaches of combining size fractionation and affinity purification to isolate in vitro-assembled splicing complexes under native conditions. Presented here is a procedure developed for isolating intact splicing complexes arrested between the first and the second chemical steps of splicing (Jurica et al., 2002).
Spliceosomes are assembled on an in vitro-transcribed splicing substrate from components present in the nuclear extract of HeLa cells. Using a substrate with a mutation at the 3' splice site, complex assembly is arrested by a block at the second step of splicing. The substrate also contains binding sites for an RNA-binding protein that serves as an affinity tag. Purification of arrested spliceosomes is carried out in three steps: (1) RNase H digestion of excess pre-mRNA substrate, (2) size-exclusion chromatography, and (3) affinity selection via the RNA-binding adapter protein (Fig. 1). Digestion serves to improve the ratio of fully assembled complexes to earlier species by targeting substrate molecules not fully assembled into spliceosomes. Cleavage is mediated by endogenous RNase H present in nuclear extracts and DNA oligonucleotides targeted to the last 30 nucleotides of the 5' exon in the pre-mRNA substrate, a region protected from digestion in arrested complexes, but accessible in earlier ones (Reichert et al., 2002). Size exclusion employs a small sizing column to quickly separate a splicing reaction into basically two fractions: larger molecules, including the spliceosomes, that are excluded by the sizing resin and elute in the void volume, and smaller molecules that can enter the pores of the resin and are retained on the column, such as the bulk of nuclear extract proteins, degraded RNA, and unbound affinity adapter protein. Affinity selection is mediated by binding sites for the bateriophage MS2 coat protein in the pre-mRNA substrate. The substrate can then be bound to a fusion of MS2 and maltose-binding protein (MBP). The MS2:MBP serves as an affinity adapter so that spliceosomes assembled on the tagged substrate can be captured by amylose resin and eluted under native conditions with maltose. The main objectives in developing this protocol were to isolate homogeneous splicing complexes in the quantities (~100µl) and concentrations (5-50nM) useful for both biochemical characterization and electron microscopy analysis.
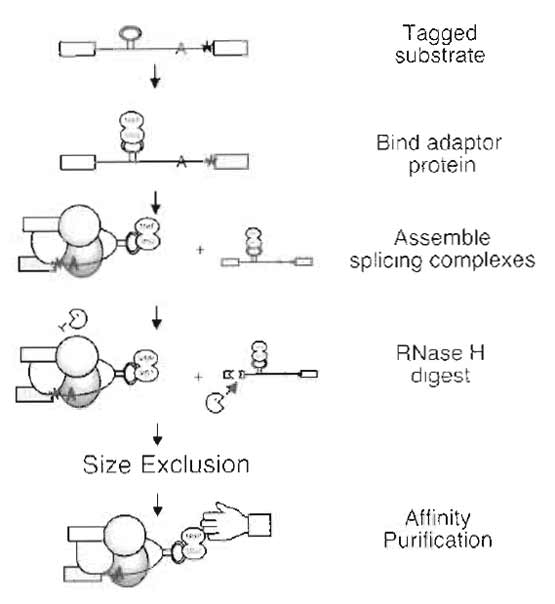 |
| FIGURE 1 Purification scheme. |
II. MATERIALS AND INSTRUMENTATION
HEPES (Cat. No. H-3375), EDTA (Cat. No. E-5134), isopropyl-β-D-thiogalactoside (IPTG, Cat. No. 1-5502), maltose (Cat. No. M-5895), phenylmethylsulfonyl flouoride (PMSF, Cat. No. P-7626), L-glutamic acid, monopotassium salt (Cat. No. G-1501), Nonidet P-40 (Cat. No. N-6507), heparin (Cat. No. H-3900), phosphocreatine (Cat. No. P-7936), yeast tRNA (Cat. No. R- 9001), Trizma base (Cat. No. T-1503), and formamide (Cat. No. 47671) are from Sigma. Tris-HCl (Cat. No. BP153), glycerol (Cat. No. G33), magnesium acetate (Cat. No. M13), and potassium chloride (Cat. No. P217) are from Fisher. Adenosine triphosphate (Cat. No. 27- 2056-01), HiTrap heparin column (Cat. No. 17-0407- 01), and S-400 Sephacryl (Cat. No. 217-0609-10) are from Amersham Biosciences. RNasin (Cat. No. N2515) is from Promega, and amylose resin (Cat. No. E8021) is from New England Biolabs. Acrylamide (Cat. No. EC-852) is from National Diagnostics, and urea (Cat. No. 821527) is from ICN. The splicing substrate is generated by in vitro runoff transcription under standard conditions (Moore and Query, 1998) from construct MJM273. MJM273 is derived from an AdML gene construct containing a single intron with an extended polypyrimidine tract, an AG->GG 3' splice site mutation (Anderson and Moore, 1997; Gozani et al., 1994), and contains a tag of three MS2-binding sites located in the intron between the 5' splice site and the branch point (Jurica et al., 2002). The substrate is transcribed with T7 RNA polymerase, radiolabeled uniformly using [α-32P]UTP and capped with G(5')ppp(5')G. The RNA should be gel purified and quantified by comparison with the specific activity of the transcription reaction. Nuclear extract is derived from HeLa cells as outlined in Dignam et al. (1983) and stored in 400-µl aliquots at -80°C. DNA oligonucleotides for RNase H-mediated digestion were synthesized with the following sequences: oligol (5'-AGCTGGCCCTCG-3'), oligo2 (5'-CAGACAGCGATG-3') and stored in 100 µM aliquots of 100 µl at -20°C.
The FPLC (Cat. No. 18-1118-67) is from Amersham Biosciences, and the Centricon-10 (Cat. No. 4205) and Kontes Flex-column (Cat. No. K420401-1010) are from Fisher. Mobicols (Cat. No. M1002) and filter (Cat. No. M2135) are obtained from Mo Bi Tec, LLC, and lowadhesion microcentrifuge tubes (Cat. No. 1415-2600) are from USA Scientific. Gel plates, combs, spacers, and electrophoresis rigs are homemade. Power supplies are from Thermo EC (EC3000) or similar, and the phosphorimager is from Molecular Dynamics.
III. PROCEDURES
A. Purification of Spliceosomes
Solutions
- Potassium glutamate: 1M, pH 7.5. To make 50ml, dissolve 9.26 g of L-glutamic acid, monopotassium salt in 35ml of H2O, and adjust the pH to 7.5 with 10N potassium hydroxide. Complete to 50 ml with H2O and filter sterilize. Store at 4°C.
- Magnesium acetate: 1M pH 7.5. To make 50ml, dissolve 10.72 g of magnesium acetate in 35 ml of H2O and adjust pH to 7.5 with sodium hydroxide. Complete to 50ml with H2O and filter sterilize. Store at room temperature.
- Creatine phosphate: 250mM. To make 5ml, dissolve 319mg phosphocreatine disodium salt in 5ml of H2O. Store at -20°C in 1-ml aliquots. Keep a 100-µl working aliquot at -20°C.
- Yeast tRNA: 5 mg/ml. Add 5 ml of H2O to a 500 unit bottle of Sigma yeast tRNA and store at -20°C. Keep a 100-µl working aliquot at -20°C.
- Heparin: 10mg/ml. Make a 1-ml stock by dissolving 10mg heparin in 1 ml H2O and filter sterilize. Store at 4°C.
- Maltose: 0.5M. To make 50ml, dissolve 9g of maltose in 40ml of H2O and complete to 50ml. Filter sterilize and store at room temperature.
- 5x SB: 0.75M KCl, 25 mM EDTA, 100mM Tris, pH 7.9. Make 50ml with 18.75ml of 2M KCl stock solution, 2.5 ml of 0.5M of EDTA stock solution, 5 ml of 1M Tris, pH 7.9, stock solution, and 23.75 ml of H2O. Store at room temperature.
- SB-N: 150mM KCl, 20mM Tris, pH 7.9, 5mM EDTA, 1 mM dithiothreitol (DTT), and 0.5% NP-40. Make 50ml fresh for each preparation with 10ml of 5 × SB, 50 µl of 1M DTT stock solution, 250 µl of 10% NP- 40 stock solution, and 39.7ml of H2O. Keep at room temperature.
- SB: 150mM KCl, 20mM Tris, pH 7.9, 5mM EDTA, and 1 mM DTT. Make 15ml fresh for each preparation with 3 ml of 5 × SB, 15 µl of 1M DTT stock solution, and 12ml of H2O. Keep on ice.
- SB-M: 150mM KCl, 20mM Tris, pH 7.9, 5mM EDTA, 1 mM DTT, and 10 mM maltose. Make 1 ml fresh for each preparation with 200µl of 5 × SB, 1 µl of 1M DTT stock solution, 20µl of 0.5M maltose, and 779 µl of H2O. Keep on ice.
Steps
- At room temperature, pour 5 ml of S-400 resin equilibrated and resuspended in an equal volume of SB-N into a 1.0 × 10-cm glass column. Allow the resin to settle by gravity flow until the buffer is near the top of the column bed. This sizing column can be used multiple times by washing with 10ml of SB-N before each use (Fig. 2A).
- Set up a 1-ml splicing reaction as described in steps 3 and 4 to contain final concentrations of the following: 10nM splicing substrate (MJM273), 500nM MS2-MBP, 80mM potassium glutamate, pH 7.5, 2mM magnesium acetate, pH 7.5, 2 mM ATP, 5 mMcreatine phosphate, 0.05mg/ml yeast tRNA, 1% RNasin, and 40% nuclear extract.
- First, place 50 µl substrate (0.2 pmol/µl) in a lowadhesion microcentrifuge tube. Heat at 95°C for 1 min and then place on ice to cool. Add 10µl MS2-MBP (50µM) and mix by flicking. Incubate on ice for 5 min.
- In a second tube, add the following in order: 381 µl H2O, 80µl potassium glutamate (1M, pH 7.5), 2 µl magnesium acetate (1M, pH 7.5), 20µl ATP (100 mM), 20 µl creatine phosphate (250 mM), 10 µl tRNA (5mg/ml, yeast), 10µl RNasin, contents of the first tube (substrate plus MS2-MBP), and 400µl nuclear extract. Mix by flicking the tube gently. (Place a 5-µl aliquot of the reaction on ice as a 0' time point for RNA denaturing gel analysis.)
- Aliquot 200µl into five tubes. Incubate tubes at 30°C for 60min to assemble spliceosomes. (Place a 5- µl aliquot of the reaction on ice as a 60' time point for RNA denaturing gel analysis.)
- During this incubation, assemble the MoBiCol column by placing a small filter in the bottom of the column. The column can be supported in a microcentrifuge tube on ice. Add 200µl of amylose resin equilibrated and resuspended in an equal volume of SB-N. Allow the buffer to drain by gravity flow (Fig. 3A).
- After the 60' incubation add 2µl each of oligo 1 and oligo 2 (100 µM) to each tube of the splicing reaction. Mix by flicking the tube gently. Incubate tubes at 30°C for 20min to induce RNase H digestion. (Place a 5-µl aliquot of the reaction on ice as an 80' time point for RNA denaturing gel analysis.)
- Add 10µl of heparin (10mg/ml) to each tube. Mix by flicking the tubes gently. Incubate tubes at 30°C for 5 min.
- Run the sizing column by gravity flow until the buffer head just reaches the top of the resin bed. Pool the splicing reactions from the five tubes and carefully load onto the sizing column. Run the column until the resin bed is exposed and load 500 µl SB-N to wash the column wall. Again run the column until the resin bed is exposed.
- Run 10 ml of SB-N through the column, taking 500-µl fractions in tubes on ice. Gravity flow should be about ~0.4 ml/min. Collect 18-20 fractions total.
- Count the fractions for the radioactively labeled the splicing substrate with a Geiger counter. There should be two peaks of radioactivity, with the first smaller peak (usually within the first 11 fractions) containing splicing intermediates (spliceosomes) and the larger second peak containing degraded splicing substrate (Fig. 2B). (This can be checked by taking 10-µl aliquots of the fractions for RNA denaturing gel analysis.)
- Pool the first peak (usually ~2ml) and flow this through the amylose column by gravity. Collect the column flow-through in tubes on ice and reapply to the amylose column. To wash the column, attach a 10-ml syringe barrel to the top of the column using a luer adaptor cap and place in a 15-ml Falcon tube. Fill the syringe with 10ml of cold SB to wash the column by gravity flow at 4°C (Fig. 3B).
- Elute the splicing complexes by pipetting 50-µl aliquots of SB-M onto the amylose resin and taking fractions dropwise into tubes on ice. Count the fractions for the radioactively labeled splicing substrate with a Geiger counter. The complexes should be contained within the first 100 µl of elution.
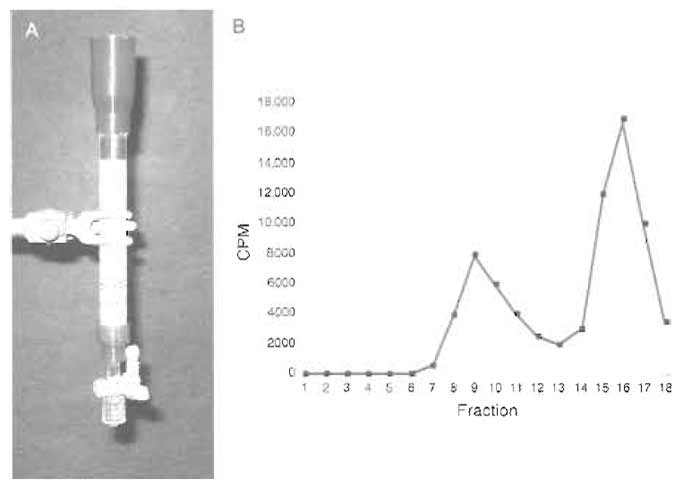 |
| FIGURE 2 (A) Size-exclusion column. (B) Elution profile of size-exclusion column measured with a Gieger counter. |
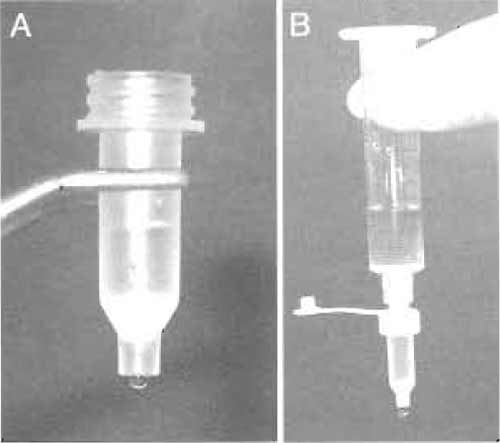 |
| FIGURE 3 (A) Amylose affinity resin in a small MoBiCol column. (B) Setup for washing the amylose column. |
B. Purifying Affinity-Tag Adapter Protein, MS2-MBP
The affinity-tag adapter protein is a recombinant MS2-MBP fusion expressed in Escherichia coli (construct a gift from Josep Vilardell). This fusion places MBP N-terminal to MS2. The MS2 portion carries a double mutation (V75Q and A81G) that prevents oligomerization (LeCuyer et al., 1995). Single-step purification of MS2-MBP over an amylose column yields a single band on a Coomassie-stained gel, but the A280/A260 ratio (<1) reveals that a significant amount of bound nucleic acid remains as a contaminant. Heparin chromatography as a second purification step eliminates this contaminant.
Solutions
- AB1: 20mM HEPES, pH 7.9, 200mM KCl, and 1 mM EDTA. Make 500ml by combining 10ml of 1M HEPES, pH 7.9, stock solution, 50ml of 2M KCl stock solution, 1 ml of 0.5 M EDTA stock solution, and 439 ml of H2O.
- AB2: 20mM HEPES, pH 7.9, 5mM KCl, and 1 mM EDTA. Make 500 ml by combining 10 ml of 1M HEPES, pH 7.9, stock solution, 1.25ml of 2M KCl stock solution, 1 ml of 0.5 M EDTA stock solution, and 487.75 ml of H2O.
- ABE: 20mM HEPES, pH 7.9, 5mM KCl, 1 mM EDTA, and 10 mM maltose. Make 100 ml by combining 2ml of 1M HEPES, pH 7.9, stock solution, 0.25 ml of 2M KCl stock solution, 200µl of 0.5M EDTA stock solution, 2 ml of 0.5 M maltose, and 474.75 ml of H2O.
- PMSF: 100mM. To make 5ml, dissolve 87.1mg PMSF in 5 ml of ethanol. Store at 4°C.
- IPTG: 1M. To make 10ml, dissolve 1.19g of IPTG in 5 ml of H2O. Store in 1-ml aliquots at -20°C.
- HB1: 20mM HEPES, pH 7.9, and 1 mM EDTA. Make 500ml by combining 10ml of 1M HEPES, pH 7.9, stock solution, 1 ml of 0.5 M EDTA stock solution, and 489ml of H2O.
- HB2: 20mM HEPES, pH 7.9, 1M KCl, and 1 mM EDTA. Make 500 ml by combining 10 ml of 1M HEPES, pH 7.9, stock solution, 250ml of 2M KCl stock solution, 1 ml of 0.5 M EDTA stock solution, and 239ml of H2O.
Steps
- Inoculate a 5-ml culture of Luria broth (LB) with single bacterial colony of DH5oc cells transformed with a plasmid expressing MS2-MBP and grow overnight to saturation at 37°C with shaking. The next morning, inoculate 1 liter of LB plus 2% glucose with the 5-ml culture. Grow the cells at 37°C with shaking to an OD600 of ~0.5 and then induce expression of the protein by adding 1 ml of 1M IPTG. Continue to grow the cells for 2-3 h and harvest by centrifugation at 6000rpm for 10min. Pour off the supernatant, freeze the cell pellet, and store at -20°C.
- Thaw and resuspend ~1 g cells in 10ml cold AB1 plus 200 µl PMSE Break open the cells by sonication on ice. Centrifuge for 30min at 15,000rpm at 4°C.
- Perform all the following steps of purification at 4°C. Load the supernatant on a -5-ml amylose column equilibrated with AB1, running the column at 0.3 ml/ min. Wash the column with 40ml of AB1, followed by 10 ml of AB2 to lower the salt concentration in preparation for heparin chromatography.
- Elute the protein with 20ml of ABE, taking 1-ml fractions. Check the OD280 of the fractions and pool the peak fractions. (The column can be cleaned with 5 ml of 0.1% SDS and reequilibrated with AB1 for future use.)
- Concentrate the pooled peak fraction to -1 ml in a Centricon -30.
- On an FPLC, equilibrate a 1-ml heparin column with a mixture of HB1 and HB2 to 5 mM KCl. Load the concentrate on the column and wash with 5 ml at 5 mM KCl.
- Run a gradient from 5 to 400mM KCl over 10 column volumes. The MS2-MBP protein elutes at ~60mM KCl. Pool peak fractions and concentrate to ~500µl in a Centricon-30. Add glycerol to 10% and freeze at -20°C in 100-µl aliquots.
- Determine the protein concentration for MS2- MBP. An OD280 of 1 corresponds to 16.5µM or 0.89mg/ml.
C. Denaturing Gel Analysis and Quantification
Solutions
- Splicing dilution buffer: 100mM Tris, pH 7.5, 10mM EDTA, 1% SDS, 150mM NaCl, and 0.3 M NaAc, pH 5.2. To make 50 ml, combine stock solutions of 5 ml of 1M Tris, pH 7.5, 1 ml of 0.5M EDTA, 5 ml of 10% SDS, 3.75 ml of 2M NaCl, 5 ml of 3M NaAc, pH 5.2, and 30.25 ml of H2O. Store at room temperature.
- Acrylamide solution: 15% acrylamide (29:1 acrylamide: bis-acrylamide), 8M urea, and 1 × TBE. To make 500ml, dissolve 240.24g of urea in 100ml of stock solution of 5 × TBE and 187.5ml of 40% acrylamide solution with stirring over low heat. Complete to 500 ml with H2O and filter. Store in the dark at room temperature.
- FEB: 94% formamide, 20mM EDTA, 0.01% cyan blue, and 0.01% bromphenol blue. To make 10ml, combine 9.4ml of formamide, 0.4ml of 0.5M EDTA, 0.1 ml of 1% cyan blue, and 0.1 ml of 1% bromphenol blue stock solutions. Store 1-ml aliquots at -20°C.
Steps
- To prepare the time point aliquots taken during the spliceosome purification for electrophoresis, remove the tubes from ice and add 95µl of splicing dilution buffer. Then add 100 µl of phenol:chloroform: isoamyl alcohol (25:24:1, pH 4.5) stock solution. Vortex well and spin at 14,000rpm in a microcentrifuge for 10min at room temperature. (If taken, aliquots from the sizing column elution should be treated similarly.)
- Take the top 90µl, avoiding the interface, and place in a new tube. To this add 3 volumes ethanol and invert to mix. Place at -70°C for 30min. Discard the lower layer.
- Spin tubes in a microcentrifuge at 14,000 rpm for 30 min at 4°C. Remove the ethanol with a pipettor and wash the pellet with 100µl of 70% ethanol. Dry the pellet.
- Resuspend the pellet in 5 µl of FEB. Load 2.5µl on the denaturing acrylamide gel.
- To prepare elution samples, add 4µl of FEB to 1 µl of elution aliquots. Load 2.5 µl on the gel.
- To prepare standard, add 39µl of FEB to 1 µl of 10nM splicing substrate used during the purification. Load 1 µl on the gel.
- Assemble a gel mold using 20 × 25-cm glass plates with 0.1mm spacers and comb. Pour the gel using 25 ml acrylamide solution and polymerize with 75 µl of 20% APS and 25 µl of TEMED stock solutions. Allow the gel to set for at least 30min.
- Assemble the gel on a gel rig with TBE buffer stock. Prerun at 30 W for 30min.
- Prior to loading on the gel, incubate the samples at 95°C for 1 min and then place on ice.
- After rinsing out the wells, load samples and standard. Run the gel at 30W for 2h. The cyan blue should migrate to near the bottom of the gel.
- Take down the gel onto a piece of exposed X-ray film and cover with plastic wrap. Expose to a phosphorimager plate overnight.
- To quantify the concentrations of the purified spliceosomes, the specific activity of the labeled splicing intermediates should be compared to the standard. (Figure 4)
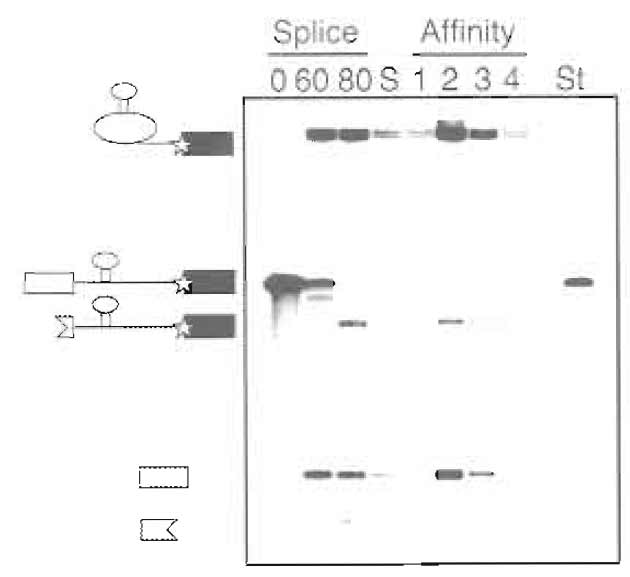 |
| FIGURE 4 Example of denaturing gel (15% polyacrylamide) analysis of substrate RNA during purification. Time points taken during the splicing reaction (0 and 60min) and after RNase H digestion (80min), size-exclusion peak fraction (S), affinity column elutions (1-4), and standard used for quantification (St) are shown. The positions of MS2-tagged pre-mRNA, lariat intermediate, 5' exon, and RNase H digestions products are indicated.. |
IV. PITFALLS
- Starting with 10 pmol of pre-mRNA substrate, this protocol generally yields 0.3-0.5 pmol of purified complexes. The yield is highly dependent on the efficiency of the first step of splicing in the reactions. Nuclear extracts can differ in their splicing efficiencies, and therefore titrations of potassium glutamate and magnesium acetate concentrations should be performed to maximize splicing efficiency for a given nuclear extract preparation.
- The final buffer conditions for the purification were chosen with electron microscopy studies in mind. We found that the splicing complexes tended to aggregate when 2mM MgCl2 was present in the buffer instead of the 5 mM EDTA. Although some spliceosome proteins are disassociated in the presence of EDTA, the core splicing components remain intact (Jurica et al., 2002).
- We have yet to find a method to concentrate spliceosomes after purification, as the complexes adhere to the membranes of all concentration devices tested. By using the very small amount of affinity resin with a column geometry, the spliceosomes can be eluted at a high enough concentration for structural and biochemical analyses without further concentration. Typically peak elution fractions of 15-50µl containing 5 to 15 nM splicing intermediates are obtained.
- Contamination of reagents, tubes, and so on by RNases is always a concern when handling RNA. Care should be taken to wear clean gloves during the purification and, if possible, to designate reagents, tips, tubes, and so on as being for RNA use only.
References
Anderson, K., and Moore, M. J. (1997). Bimolecular exon ligation by the human spliceosome. Science 276, 1712-1716.
Das, R., Zhou, Z., and Reed, R. (2000). Functional association of U2 snRNP with the ATP-independent spliceosomal complex E. Mol. Cell 5, 779-787.
Dignam, J. D., Lebovitz, R. M., and Roeder, R. D. (1983). Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11,1475-1489.
Gozani, O., Patton, J. G., and Reed, R. (1994). A novel set of spliceosome- associated proteins and the essential splicing factor PSF bind stably to pre-mRNA prior to catalytic step II of the splicing reaction. EMBO J. 13, 3356-3367.
Hartmuth, K., Urlaub, H., Vornlocher, H. P., Will, C. L., Gentzel, M., Wilm, M., and Luhrmann, R. (2002). Protein composition of human prespliceosomes isolated by a tobramycin affinityselection method. Proc. Natl. Acad. Sci. USA 99, 16719-16724.
Jurica, M., Licklider, L., Gygi, S., Grigorieff, N., and Moore, M. (2002). Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 8, 426-439.
Jurica, M. S., and Moore, M. J. (2002). Capturing splicing complexes to study structure and mechanism. Methods 28, 336-345.
LeCuyer, K. A., Gehlen, L. S., and Uhlenbeck, O. C. (1995). Mutants of the bacteriophage MS2 coat protein that alter its cooperative binding to RNA. Biochemistry 34, 10600-10606.
Makarov, E. M., Makarova, O. V., Urlaub, H., Gentzel, M., Will, C. L., Wilm, M., and Luhrmann, R. (2002). Small nuclear ribonucleoprotein remodeling during catalytic activation of the spliceosome. Science 298, 2205-2208.
Moore, M. J., and Query, C. C. (1998). Uses of site-specifically modified RNAs constructed by RNA ligation. In "RNA-Protein Interactions: A Practical Approach" (C. W. J. Smith, ed.), pp. 75-108. IRL Press, Oxford.
Moore, M. J., Query, C. C., and Sharp, P. A. (1993). Splicing of precursors to mRNA by the spliceosome. In "The RNA World" (R. Gesteland and J. Atkins, eds.), pp. 303-357. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Reichert, V., Le Hir, H., Jurica, M. S., and Moore, M. J. (2002). 5' exon interactions within the human spliceosome establish a framework for exon junction complex structure and assembly. Genes Dev. 16, 2778-2791.
Zhou, Z., Sim, J., Griffith, J., and Reed, R. (2002). Purification and electron microscopic visualization of functional human spliceosomes. Proc. Natl. Acad. Sci. USA 99, 12203-12207.
Support our developers




