Purification of Rat Liver Golgi Stacks
I. INTRODUCTIONThe study of intracellular organelles has been facilitated greatly by their purification from cellular homogenates. Such protocols yield an abundant source of material for both structural and functional studies. This article describes some simple protocols derived from several earlier methods (Leelavathi et al., 1970; Fleischer and Fleischer, 1970; Hino et al., 1978) for oµaining highly purified Golgi stacks from rat liver and for determining their relative purity over the homogenate.
II. MATERIAL AND INSTRUMENTATION
Centrifugation is carried out using an L8-70M or Optima LE-80K ultracentrifuge and either a SW-28 rotor (Cat. No. 342204) containing ultraclear tubes (Cat. No. 344058) or a SW-41 rotor (Cat. No. 331336) containing ultraclear tubes (Cat. No. 344059) from Beckman Coulter Inc. A 150-µm-mesh stainless-steel laboratory test sieve (Cat. No. 200SBW.150) and a stainless-steel receiver (Cat. No. 200BRASREC) are from Endecotts Ltd. A 0-50% Delta refractometer (Cat. No. 2-70) is from Bellingham and Stanley Ltd. Scintillation counter LS6500, spectrophotometer DU530 is from Beckman Coulter Inc.
Potassium phosphate dibasic (Cat. No. 3252), potassium phosphate (Cat. No. 3246), sucrose (Cat. No. 4072), magnesium chloride (Cat. No. 2444), concentrated hydrochloric acid (Cat. No. 9535), and dimethyl sulfoxide (DMSO, Cat. No. 9224) are from J. T. Baker. Tris (Cat. No. AB02000), SDS (Cat. No. AB01920), and β-mercaptoethanol (Cat. No. AB01340) from American Bioanalytical. The Bio-Rad protein assay (Cat. No. 500- 0006) is from Bio-Rad. Manganese chloride (Cat. No. M-3634), Triton X-100 (Cat. No. T-9284), phosphotungstic acid (Cat. No. P-4006), ovomucoid (Cat. No. T-2011), ATP (Cat. No. A-7699), UDP-galactose (Cat. No. U-4500), pepstatin A (Cat. No. P-5318), and sodium cacodylate (Cat. No. C4945) are from Sigma. Complete EDTA-free tablet (protease inhibitor cocktail tablets, Cat. No. 1 873 580) is from Roche. [3H]UDPgalactose with a specific activity of 30-50Ci/mmol (Cat. No. NET758) is from NEN Research products. Ethyl alcohol (190 proof, ACS/USP grade) is from Pharmco products, Inc. Scintillation fluid (Opti-Fluor, Cat. No. 6013199) is from Packard BioScience. All reagents are of analytical grade or better. The water used is double distilled and filtered.
III. PROCEDURES
A. Purification of Rat Liver Golgi Stacks
Solutions
- 0.5M potassium phosphate buffer, pH 6.7: Make up 500-ml solutions of 0.5 M anhydrous K2HPO4 (43.55 g) and 0.5 M anhydrous KH2PO4 (34.02 g). To 400 ml of the latter, gradually add the former until the pH reaches 6.7. Store at 4°C.
- 2 M sucrose: Dissolve 342.3 g in water prewarmed to 50°C. Make up to a final volume of 500 ml and store at 4°C.
- 2M MgCl2: Dissolve 40.7g of MgCl2·6H2O in water to a final volume of 100ml. Store at room temperature.
- Protease inhibitors: Directly dissolve one EDTAfree tablet in 50ml solution and then add 50µl pepstatin A (5 mM stock). To prepare the stock solution of pepstatin A, dissolve 3.43mg of the powder in 1 ml DMSO and store at -20°C.
- Gradient buffers: Make up buffers A-E from the preceding three stock solutions and cold water as shown in Table I. Fifty milliliters of each buffer A, B, and E and 100ml of buffers C and D are needed for a medium-scale preparation. EDTA-free protease inhibitor tablet (1 tablet per 50 ml buffer) and pepstatin A (5µM) are added to buffer C and D. We normally make 500ml buffer C without protease inhibitors (which are expensive) for washing the liver. The water should be precooled to 4°C overnight to ensure that all the buffers are ice cold.
It is very important to be as accurate as possible when mixing various components and to check the refractive index of each buffer using a refractometer. The final refractive index should be adjusted to within ±0.5% sucrose (about 0.001 in refractive index) for buffers C and D in particular.
 |
1. Standard Protocol (Medium-Scale Preparation)
This common protocol is used for general biochemical studies of Golgi membranes. It suffices for many electron microscopy studies as well.
Steps
- Starve six female Sprague-Dawley rats (body weight 150-200 g) for 24 h.
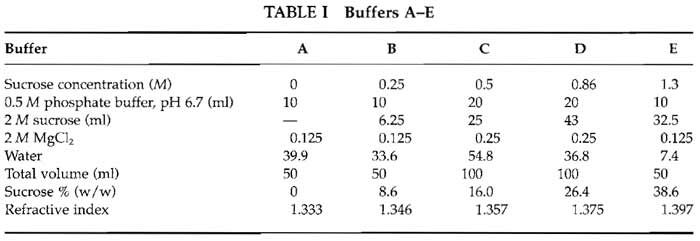
- After killing the rats with CO2, rapidly remove the livers into large volume of ice-cold buffer C without protease inhibitors. This cools the liver and washes off the blood. Then transfer the livers into icecold buffer C with protease inhibitors. Mince into small pieces (approximately 4-5 mm in diameter) with a pair of scissors (Fig. 1A). Put the livers into a 50-ml Falcon tube.
- Check the weight of livers, which is normally 40-45g. A maximum of 50g liver can be used for 12 gradients.
- Homogenize the tissue by gently pressing through a 150-µm-mesh stainless-steel sieve with the bottom of a 250-ml conical flask using a rolling action (Fig. 1B). Adding a small amount of buffer C (with protease inhibitor) to the sieve will make it easier to press the liver through the mesh (Fig. 1C). Pool the homogenate (Fig. 1D) into a 50-ml Falcon tube; the final volume should be about 50 ml. Keep 200 µl on ice for enzyme assay.
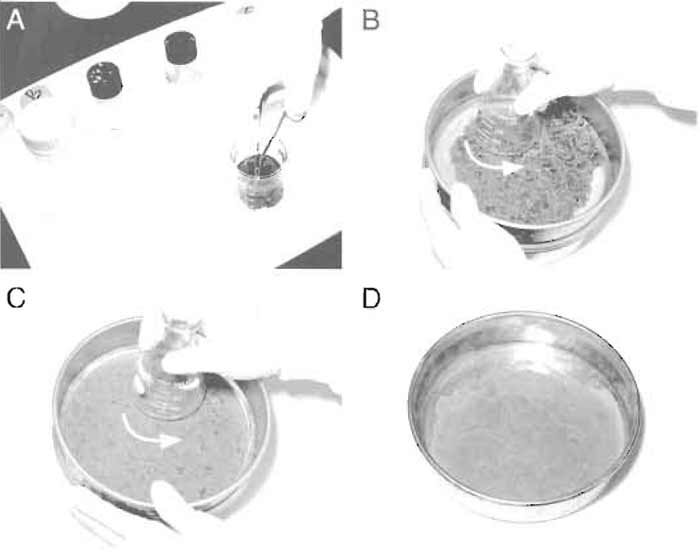
- Gradients (Fig. 2A):
- Place 6 ml of buffer D in each of the 12 SW-41 ultraclear tubes (size 14 × 89mm).
- Overlay 4.5 ml of homogenate.
- Overlay 1.8ml of buffer B and balance the tubes to within 0.01 g.
- Centrifuge at 29,000rpm in a SW-41 rotor for 60min at 4°C.
- Aspirate and discard the lipid at the top and the cytosol (colored red by hemoglobin) and collect Golgi fractions that accumulate at the 0.5/0.86M interface (cloudy band) with a plastic Pasteur pipette (Fig. 2B).
- Pool the fractions in a 50-ml Falcon tube. Typically, the total volume is about 15 ml with a refractive index of about 1.370 (0.77M sucrose). Keep 200B1 on ice for enzyme assay. Adjust the Golgi sample to 0.25M sucrose (refractive index 1.345) using buffer A. Make up to 50 ml with buffer B if necessary.
- For the second gradient (Fig. 2C), put 1 ml of buffer E followed by 2ml of buffer C into each tube and then overlay 8.0ml of the diluted Golgi fractions.
- Centrifuge at 8000rpm in a SW-41 rotor for 30min at 4°C.
- Aspirate and discard the supernatant and collect the membranes (thin band) at the 0.5M/1.3M sucrose interface (Fig. 2D). Gently mix the Golgi membranes (about 1.5-2ml) with 1 volume buffer A. Final volume is 3-4ml. Check the protein concentration by the Bradford assay (Bio-Rad); typically this is 1- 2mg/ml.
- Aliquot and freeze samples in liquid nitrogen and then store at -80°C. Samples can be thawed and frozen twice without significant loss of enzymatic activity or loss of morphology.
 |
| FIGURE 1 Steps in homogenizing rat liver. (A) Mince rat liver in ice-cold buffer C. (B and C) Gently press the liver through the metal mesh. (D) Homogenate in the sieve receiver. |
 |
| FIGURE 2 Sucrose gradients for Golgi purification from rat liver. (A) Liver homogenate was first loaded onto a 0.86M sucrose cushion (buffer D) and overlaid with 0.25 M sucrose (buffer B). Letters on the left indicate sucrose buffers used to make the gradients. (B) After ultracentrifugation, Golgi stacks are enriched at the 0.5/0.86M interface. (C) Crude Golgi sample from the first gradient was loaded onto the second gradient. (D) After centrifugation, Golgi stacks form a thin band at the 0.5/1.3M interface and can be collected using a Pasteur pipette. |
2. Large-Scale Preparation
This protocol ensures large amounts of Golgi membranes with minimal contamination by other membranes. However, stacked cisternal structures may be less well preserved. The addition of protease inhibitors during the procedure is optional (see Section IV). Two consecutive spins at high speed are used to accommodate the large amount of homogenate used.
Steps
- Use 8-12 female rats depending on size and requirement. Starve for 24h. Kill the rats and ensure that all subsequent steps are performed as quickly as possible.
- Rapidly remove the livers into 250ml of ice-cold buffer C (without protease inhibitors) while swirling to speed cooling.
- Weigh the livers, transfer to fresh buffer C, and mince the tissue into small pieces (about 5mm in diameter) with a pair of scissors (Fig. 1A).
- Homogenize the tissue by gently pressing through a 150-µm-mesh stainless-steel sieve using the base of a 250-ml conical flask (Figs. 1B and 1C). Take 100 µl of the homogenate for assay and record the total volume. The final volume for up to 100g of liver should be about 120ml, which is the maximum for six gradients.
- Gradients:
- Place 16 ml buffer D in each of the six SW-28 ultraclear tubes.
- Overlay each gradient with 18-20ml of the homogenate.
- Finally overlay with buffer B to top up and balance the tubes.
- Centrifuge at 28,000rpm in a SW-28 rotor for 60min at 4°C.
- Aspirate the lipid and collect Golgi fractions that accumulate at the 0.5/0.86 M interface between buffers C and D. About 3-4 ml should be collected from each gradient.
- Pool the fractions and dilute to 0.5M sucrose using buffer A. Check the refractive index: 0.5M sucrose reads 1.357. Record the volume and keep 100µl for enzyme assay. The total volume should be about 40ml and should not exceed 60ml.
- For the second gradient, add 2ml buffer E into SW-41 tubes and overlay 8-10 ml diluted Golgi sample. Spin at 8000rpm for 30min at 4°C.
- Aspirate and discard supernatant as before. Collect membranes at the 0.5/1.3M sucrose interface. Gently mix the Golgi sample (about 2.5ml) with 1 volume buffer A. Final volume is about 5 ml. Check the protein concentration by the Bradford assay (Bio-Rad), which should typically be about 2mg/ml.
- Aliquot and freeze samples in liquid nitrogen and store at -80°C.
B. Determination of β-1,4- Galactosyltransferase Activity
The relative purification of the Golgi stacks can be assessed by measuring the increase in specific activity of the Golgi enzyme β-l,4-galactosyltransferase (GAIT) over that of the whole liver homogenate. The enzyme assay used here is that of Bretz and Staubli (1977), which measures the addition of tritiated galactose onto the oligosaccharides of an acceptor protein, ovomucoid.
Solutions
- 0.4M sodium cacodylate, pH 6.6: Dissolve 17.1g in 150ml of water and adjust the pH to 6.6 with HCl. Make up to 200ml and store at room temperature.
- 175mg/ml ovomucoid: Dissolve 1 g in water and make up to a final volume of 5.7 ml. Filter through a 0.45-µm nitrocellulose filter, aliquot, and store at -20°C.
- 10mM UDP-galactose: Dissolve 25mg in a final volume of 4.4ml of water. Aliquot and store at -20°C.
- 10% (w/v) Triton X-100: Dissolve 10 g in 80 ml of water and make up to 100ml. Store at 4°C.
- 0.2 M ATP: Dissolve 605 mg in 3 ml of water. Adjust the pH to 6.5-7.0 with 1M NaOH and make up to a final volume of 5ml with water. Aliquot and store at -20°C.
- 2 M MnCl2: Dissolve 9.9 g of MnCl2·4H2O in 15 ml of water and make up to 25 ml. Store at 4°C.
- 1% phosphotungstic acid/0.5M HCl (PTA/HCl): Dissolve 5g of phosphotungstic acid in 400ml of water. Add 22ml of concentrated HCl and make up to 500ml with water. Store at 4°C.
- 5% (w/v) SDS: Dissolve 5g of SDS in 80ml of water and make up to 100ml. Store at 4°C.
- 2M Tris: Dissolve 24.2 g of Tris in 70ml of water and make up to 100 ml. Store at room temperature.
- Assay mixture: Make up a fresh batch of the assay mixture from the aforementioned stocks as follows: 200µl sodium cacodylate, 6µl β-mercaptoethanol, 200µl ovomucoid, 40µl UDP-galactose, 40 µl Triton X-100, 20 µl ATP, 40 µl MnCl2, 10 µl [3H]UDP-galactose, and 1040 µl of water. The concentration of UDP-galactose in the assay mixture is 0.25 mM.
Steps
- Once the Golgi preparation has been completed, make 1:20 dilutions of the homogenate, intermediate, and Golgi fractions using water.
- Add 80µl of assay mixture (in duplicate) to screwcapped Eppendorf tubes containing 20µl of the diluted samples or water (blanks). Vortex and incubate at 37°C for 30min.
- Stop the reaction by adding 1 ml of ice-cold PTA/HCl and spin at 13,000rpm on a bench-top centrifuge for 10s.
- Aspirate and discard the supernatants. Add 1 ml of PTA/HCl. Resuspend the pellets by vortexing and spin as in step 3.
- Aspirate and discard the supernatants. Add 1 ml of ice-cold 95% ethanol and resuspend the pellets as in step 4.
- Spin as in step 3 and discard the supernatant. Resuspend the pellets in 50µl of 2M Tris followed by 200 µl of 5% SDS. Shake or vortex until dissolved.
- Add 10µl of assay mixture (containing 2.5 nmol of UDP-galactose), 40 µl of water, and 200 µl of 5% SDS to a fresh tube. This allows determination of the [3H]UDP-galactose-specific activity in the mixture.
- Add 1 ml of scintillation fluid to each sample. Vortex and count in a scintillation counter using the tritium channel.
C. Determination of Protein Concentration
Steps
- While the GaIT assays are incubating, make up the following dilutions of the three samples with water: 1:100 for the homogenate, 1:20 for the intermediate fraction, and 1:2 for the Golgi fraction.
- Prepare a diluted Bio-Rad protein assay solution (fivefold dilution with water)
- Aliquot 10 µl of each of the diluted samples in duplicate into 1-ml cuvettes using 10µl water as a blank. Also aliquot 10µl of 0.1, 0.2, 0.4, and 0.8mg/ml of bovine serum albumin (BSA) for preparation of a standard curve.
- Add 990µl of the diluted Bio-Rad protein assay solution, mix well, and measure the absorbance at 595 nm.
D. Calculations of Purification Tables
Steps
- Construct a protein standard curve by plotting the
absorbance of each standard against the concentration
of BSA. Calculate the slope (m) and the intercept
at the ordinate axis (c).
Protein concentration (mg/ml) = (sample absorbance- c) × dilution m
Total protein (mg) = Protein concentration (mg/ml) × volume (ml) - Calculate the specific activity (SA) of [3H]UDPgalactose
in the assay mixture as
SA[3H]UDP-galactose (dpm/nmol) = dpm of standard- blank 2.5nmol
10 µl assay mixture contains 2.5 nmol of UDP-galactose (Section III, B, step 7). - Calculate the GalT activity in each sample as
GalT activity concentration (nmol/h/ml) = average dpm- blank × 1 × 1 × 20 (dilution factor) SA[3H]UDP-galactose 0.02 ml 0.5 hr
Total GaIT activity (nmol/h) = GaIT activity concentration (nmol/h/ml) × volume (ml) - Calculate the SA of GalT by dividing its concentration by the protein concentration of the same sample to give SA in nanomoles per hour per milligram at 37°C.
- The yields of Golgi membranes can be calculated from the ratio between the total GalT activity in the Golgi fractions and that of the homogenate.
- The purification fold is the factor by which the SA of GalT increases in the Golgi fractions over the homogenate.
IV. COMMENTS
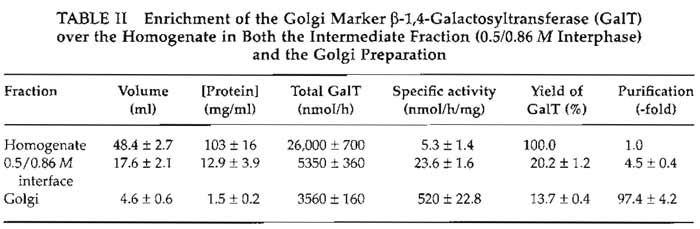
This protocol typically yields Golgi membranes that are purified 80- to 100-fold over the homogenate, as depicted in Table II. The Golgi preparations contain very little lysosomal or endoplasmic reticulum contamination as assessed by assay of β-N-acetylhexosaminidase (Landegren, 1984) and rotenone-insensitive NADH-cytochrome c reductase (Sottocasa et al., 1967). The stacked nature of these Golgi membranes can be confirmed by examination of preparations by electron microscopy (Fig. 3). Samples are fixed in suspension by the method described by Pypaert et al. (1991).
The addition of protease inhibitors, although not essential, is recommended, especially when the Golgi protein of interest is known to be sensitive to proteases. The addition of protease inhibitors can reduce the apparent purification factor, although the reasons are not clear.
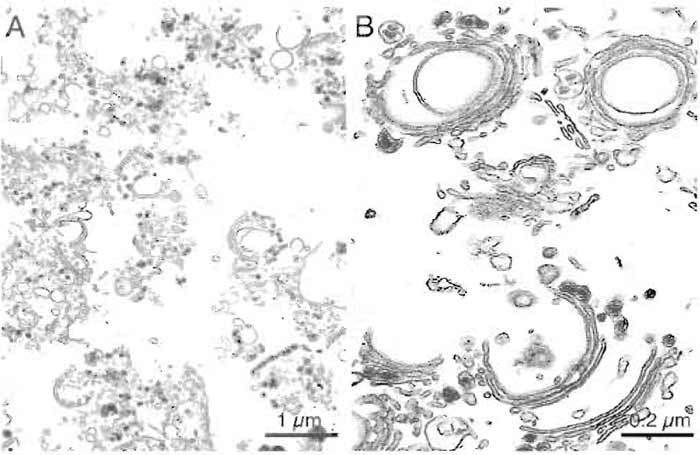 |
| FIGURE 3 Representative micrographs of a typical rat liver Golgi preparation showing stacked membranes at low (A) and high (B) magnification. Bars: 1 µm (A) and 0.2µm (B). |
 |
V. PITFALLS
- As with many organelle purification procedures, it is vital to keep all solutions at 4°C during the whole protocol to prevent excessive protease digestion. If possible, steps 3-5 should be performed in a cold room. The addition of a protease inhibitors helps limit sample proteolysis.
- All steps should be carried out as quickly as possible, and the entire procedure, from killing of the rats to freezing of the final samples, should take approximately 3-3.5 h.
- Gradients should not be overloaded by increasing the concentration of the homogenate, as this increases the amount of mitochondrial contamination.
- The final Golgi pellet should be white. A brown pellet indicates the presence of contaminating mitochondria. Lowering the concentration of the homogenate reduces such contamination.
- The 150-µm sieve will become clogged with connective tissue after excessive use. This can be removed after each preparation by soaking the sieve in 4M NaOH for 20-30min followed by washing with copious amounts of water and brushing. Prolonged soaking in 4M NaOH attacks the glue used in binding the mesh to the wall of the sieves and should therefore be avoided.
Acknowledgments
The authors thank the former and current members of the Warren laboratory for contributions to the development of these methods, Henry Tan for providing the photographs in Fig. 1 and 2, and Matthew Beard for critical reading of the manuscript.
References
Bretz, R., and Staubli, W. (1997). Detergent influence on rat-liver galactosyltransferase activities towards different acceptors. Eur. 1. Biochem. 77, 181-192.
Fleischer, B., and Fleischer, S. (1970). Preparation and characterization of Golgi membranes from rat liver. Biochem. Biophys. Acta 219, 301-319.
Hino, Y., Asano, A., Sato, R., and Shimizu, S. (1978). Biochemical studies of rat liver Golgi apparatus. I. Isolation and preliminary characterisation. J. Biochem. (Tokyo) 83, 909-923.
Landegren, U. (1984). Measurement of cell numbers by means of the endogenous enzyme hexosaminidase: Applications to the detection of lymphokines and cell surface antigens. J. Immunol. Methods 67, 379-388.
Leelavathi, D. E., Estes, L. W., Feingold, D. S., and Lombardi, B. (1970). Isolation of a Golgi-rich fraction from rat liver. Biochim. Biophys. Acta 211, 124-138.
Pypaert, M., Mundy, D., Souter, E., Labbe, J.-C., and Warren, G. (1991). Mitotic cytosol inhibits invagination of coated pits in broken mitotic cells. J. Cell Biol. 214, 1159-1166.
Sottocasa, G. L., Kuylenstierna, B., Ernster, L., and Bergstrand, A. (1967). An electron transport system associated with the outer membrane of liver mitochondrial. J. Cell Biol. 32, 415-438.
Support our developers




