Reprogramming Somatic Nuclei and Cells with Cell Extracts
I. INTRODUCTION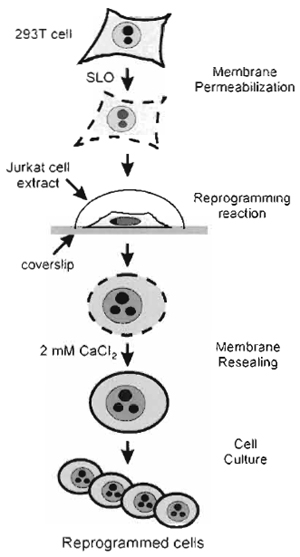 |
| FIGURE 1 Strategy of in vitro cell reprogramming. 293T cells grown on coverslips are reversibly permeabilised with SLO. The permeabilised cells are incubated in a nuclear and cytoplasmic extract derived from Jurkat TAg cells for 1 h. The cells are resealed for 2h in culture medium containing 2mM CaCl2. The CaCl2- containing medium is replaced by regular complete culture medium, and the resealed, reprogrammed cells are cultured for assessment of reprogramming. |
Nuclear reprogramming occurs in a variety of natural and experimental contexts. After fertilization, epigenetic alterations of the embryonic genome take place during successive stages of development. Epigenetic changes and alterations in gene expression also occur after the fusion of somatic cells with less differentiated cell types (Blau and Blakely, 1999; Tada et al., 2001). The birth of clones and the production of embryonic stem cells by transplantation of nuclei into oocytes also provide evidence of complete reprogramming of somatic nuclei (Cibelli et al., 1998; Gurdon et at., 1979; Munsie et al., 2000; Wilmut et al., 1997). Based on these examples, reprogramming can be defined as an alteration of a differentiated nucleus into a totipotent or mutipotent state. Additional studies have shown that a somatic cell type could be, at least partially, turned into another somatic cell type. This was achieved in coculture conditions (Morrison, 2001) and, more recently, by exposing somatic nuclei or cells to an extract derived from another somatic cell type (Håkelien et al., 2002; Landsverk et al., 2002). These observations have led to the proposal of a simple definition of nuclear reprogramming. Reprogramming may not necessarily involve dedifferentiation or return to a more pluripotent state, but may refer to the dominance of the program of one cell type over another, resulting in "the transformation of the pliant nucleus [in]to the dominant type" (Western and Surani, 2002).
We describe a procedure to redirect the nuclear program of a transformed human fibroblast cell line toward a T-cell program. The approach is outlined in Fig. 1 and may, in principle, be applied to multiple cell types. The procedure involves the use of a nuclear and cytoplasmic extract derived from Jurkat T-cells in which reversibly permeabilized fibroblasts are incubated. At the end of incubation, the fibroblasts are resealed and cultured to assess the expression of T-cellspecific markers and the establishment of T-cellspecific functions. As large numbers of cells or nuclei can be processed simultaneously, and considering the ability of cell extracts to be manipulated biochemically, in vitro manipulation of nuclei and cells provides a potentially powerful system for analyzing the mechanisms of nuclear reprogramming.
II. MATERIALS AND INSTRUMENTATION
A. Materials
- 293T human fibroblasts cultured on glass coverslips
- Round, 12-mm glass coverslips, autoclaved
- Poly-L-lysine (Cat. No. P8920, Sigma-Aldrich Co., St. Louis, MO)
- Propidium iodide (Cat. No. P4170, Sigma). Make a 1-mg/ml stock solution in H2O and store at -20°C in the dark.
- RPMI 1640 medium (Cat. No. R0883, Sigma) supplemented with 10% fetal calf serum
- Hanks balanced salt solution (HBSS; Cat. No. 14170-088, Gibco-BRL; Paisley, UK)
- Protease inhibitor cocktail (Cat. No. P2714, Sigma). This is a 100× stock solution.
- Cell lysis buffer (50 mM NaCl, 5 mM MgCl2, 20 mM HEPES, pH 8.2, l mM dithiothreitol, 0.1mM phenylmethylsulphonyl fluoride and the protease inhibitor cocktail) at 4°C
- Streptolysin O (SLO) (Cat. No. S5265, Sigma) at 100µg/ml in H2O, aliquoted, and stored at -20°C
- 1M CaCl2 (Cat. No. C4901, Sigma) in sterile H2O
- ATP (Cat. No. A3377, Sigma) at 200mM in H2O, aliquoted, and stored at -20°C
- Creatine kinase (Cat. No. C3755, Sigma) at 5 mg/ml in H2O, aliquoted, and stored at -20°C
- Phosphocreatine (Cat. No. P7936, Sigma) at 2M in H2O, aliquoted, and stored at -20°C
- GTP (Cat. No. G8752, Sigma) at 10mM in H2O, aliquoted, and stored at -20°C
- Nucleotide triphosphate (NTP) set (Cat. No. 1277057, Roche; Basel, Switzerland). Prepare a stock solution by mixing 20µl of each NTP provided in the set at a 1:1:1:1 ratio on ice. Aliquot in 10 µl and store at-20°C This makes an NTP mix at 25 mM of each NTP. Prepare more stock solution as needed.
B. Instrumentation
- Sonicator fitted with a 2-mm-diameter probe (Model Labsonic M, B. Braun Biotech International; Melsungen, Germany)
- Regular atmosphere incubator set at 37°C (for human cells)
- 5% CO2 incubator set at 37°C
- 50- and 15-ml conical tubes (Corning; Corning, NY)
- 1.5-ml centrifuge tubes
- 24-well cell culture plates (Costar Cat. No. 3524, Corning)
- Refrigerated centrifuge with swinging buckets suited for 15- and 50-ml conical tubes
III. PROCEDURES
The methods describe (1) the preparation of "donor" cells to be reprogrammed, (2) the preparation of the reprogramming extract, (3) the permeabilisation of the donor cells, (4) the reprogramming reaction, (5) the resealing of the reprogrammed cells, and (6) examples of assessments of nuclear and cell reprogramming. The procedures described are based on the reprogramming of a human fibroblast cell line (293T) in an extract derived from the human Jurkat TAg cell line (Håkelien et al., 2002).
A. Seeding 293T Cells
On the day prior to reprogramming reaction, plate 293T cells onto 12-mm round, sterile, poly-L-lysinecoated glass coverslips at a density of 50,000 cells per coverslip. Each coverslip is placed in individual wells of a 24-well culture plate. Overlay coverslips with 500µl of complete RPMI 1640 medium and place in a 5% CO2 incubator at 37°C.
B. Preparation of the Reprogramming Extract
1. Cell Harvest
- Transfer the Jurkat TAg cell suspension culture into 50-ml conical tubes and sediment the cells at 800g for 10 min at 4°C.
- Wash the cells twice in ice-cold phosphate-buffered saline (PBS) by suspension and sedimentation at 800g for 10min at 4°C. Cells can be pooled into a single tube after the first wash.
2. Cell Swelling
- Resuspend the cells in 10 ml ice-cold cell lysis buffer (CLB). It is preferable to use a graduated 15-ml conical tube to estimate the cell volume after sedimentation.
- Centrifuge at 800g for 10min at 4°C.
- Estimate the volume of the cell pellet. Resuspend the pellet into 2 volumes of ice-cold CLB.
- Hold the cells on ice for 45 min to allow swelling. This makes it easier to lyse the cells during sonication. Keep the cells well suspended by occasional tapping of the tube during swelling. Note that the cells can be allowed to swell for longer than 45 min. This swelling step can be omitted for Jurkat TAg or primary T-cells as these cell types lyse promptly during sonication.
3. Extract Preparation
- Aliquot the cell suspension into 200µl in 1.5-ml centrifuge tubes previously chilled on ice. Sonicate each tube one by one (on ice) until all cells and nuclei are lysed. Lysis is assessed by complete disruption of the cells and nuclei, as judged by the sole appearance of cell "debris" by phase-contrast microscopy examination of 3-µl samples. Once lysis is achieved in a tube, keep the tube on ice and proceed with all other tubes. Power and duration of sonication varies with each cell type. For Jurkat TAg cells, sonication of each tube at 25% power and 0.5-s pulse cycle over 1rain 40sec is recommended.
- Pool all the lysates into one (or multiple, if needed) chilled 1.5-ml centrifuge tube. Sediment the lysate at 15,000g for 15rain at 4°C in a fixedangled rotor. Note that a swing-out rotor can also be used.
- Carefully collect the supernatant with a 200-µl pipette and transfer it into a new 1.5-ml tube chilled on ice. This is the reprogramming extract.
- It is possible to aliquot the extract into 200-µl tubes such as those used for polymerase chain reaction, with 100 µl extract per tube. Snap-freeze each tube in liquid N2 and store at -80°C. However, we recommend carrying out reprogramming with freshly made extract as the stability of the extract at -80°C may vary with cell types and batches.
- Following sedimentation in step 3, remove 20µl of extract to determine protein concentration and pH. The protein concentration should be between 20 and 25mg/ml. The pH should be between 6.7 and 7.0 (see Comment 2).
4. Extract Toxicity Assay
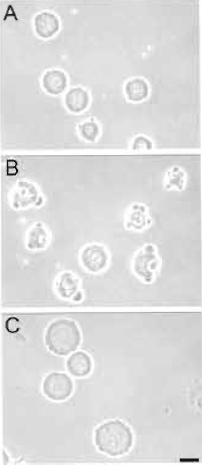 |
| FIGURE 2 Results from toxicity assay. Rat embryo fibroblasts were exposed to a eprogramming extract for 1 h as described in the text. Cells were examined by phase-contrast microscopy. (A) Viable cells. (B) Nonviable cells. Reprogramming extracts producing such cells are discarded. (C) Control cells exposed for 30min to the cell lysis buffer used to prepare the extract. Bar: 20 µm. |
Each new batch of Jurkat TAg cell extract requires a cell toxicity test.
- Add 50,000 293T cells (or, in principle, HeLa cells or any other epithelial of fibroblast cell line growing in the laboratory) to 35 µl of extract on ice in a 1.5-ml centrifuge tube. The extract does not need to contain any additives (unlike for a reprogramming reaction; see Section III,E,1).
- Incubate for 1 h at 37°C in a water bath.
- Remove a 3-µl aliquot and assess cell morphology by phase-contrast microscopy. Fig. 2 illustrates primary rat fetal fibroblasts after a 30-min exposure to reprogramming extracts. In our hands, the morphology of the cells after a 30-min incubation in the extract reflects their survival in culture as judged 24 h after the toxicity assay. Cells shown in Fig. 2A survive the extract exposure, whereas cells in Fig. 2B have been damaged by the extract and do not survive in culture. Extract batches producing such cells should be discarded.
- If so wished, replate the cells directly from the extract in complete RPMI 1640 for an overnight culture to assess survival further. There is no need to remove the extract prior to replating.
C. Permeabilisation of 293T Cells
In order for components from the reprogramming extract to enter 293T cells, the cells must be reversibly permeabilised. Permeabilisation is accomplished with the Streptococcus pyogenes toxin, streptolysin O. SLO is a cholesterol-binding toxin that forms large pores in the plasma membrane of mammalian cells (Walev et al., 2002).
1. Preparation of SLO Stock Solution
- Dissolve SLO powder in sterile-filtered MilliQ H2O to 100 µg/ml. Keep on ice while dissolving the SLO.
- Aliquot 10µl in 200-µl tubes and store at-20°C
- Discard all tubes after 1 month of storage at-20°C and prepare a new stock. Stock aliquots should be thawed only once. Note that because commercial batches of SLO vary in specific activity, a range of SLO concentrations (200, 500, 1000, 2000, and 4000ng/ml SLO) should be tested on the cell type to be reprogrammed after a new stock is prepared.
2. Cell Permeabilisation
- Dilute the SLO stock in ice-cold HBSS to 230ng/ml. This is the working solution. Note that this concentration is valid for 293T cells and should be adjusted for other cell types using a cell permeabilisation assay described in Section III,D.
- Keep the SLO on ice until addition to 293T cells.
- Remove the RPMI 1640 medium from wells containing 293T cells grown on coverslips and wash the cells four times with PBS at room temperature to remove all Ca2+ from the culture medium. This step is essential as Ca2+ inhibits SLO activity.
- Add 250µl SLO working solution to each well.
- Incubate at 37°C in regular atmosphere for 50min. Proceed to Section III,E,1.
D. Cell Permeabilisation Assay
This assay allows evaluation of the efficiency of the SLO treatment. It is recommended to carry out this assay using four additional coverslips supporting 293T cells as described in Section III,A, in addition to those used for the reprogramming reaction. This assay is based on the uptake of the fluorescent DNA stain, propidium iodide, by permeabilised cells, but not by intact cells.
- Permeabilise cells on two coverslips with SLO as described under Section III,C,2. However, in the first step, add propidium iodide to 0.1 µg/ml to the SLO dilution in HBSS on one of the coverslips. Propidium iodide will be taken up as cells are being permeabilised. The other coverslip receives 250µl SLO dilution in HBSS without propidium iodide.
- Two additional coverslips should also be used as controls for the absence of SLO. Add 250µl HBSS containing propidium iodide to one of the control coverslips as in step 1. The other coverslip receives 250µl HBSS without propidium iodide.
- Incubate at 37°C in regular atmosphere for 50 min.
- For SLO-treated and control coverslips not containing propidium iodide, remove HBSS and immediately add 1.5ml of preheated (37°C) complete RPMI 1640 containing 2 mM Ca2+ added from a 1M stock (see Section II,A). Incubate at 37°C for 2h to allow resealing of the plasma membranes.
- For SLO-treated and control coverslips labelled with propidium iodide, remove HBSS, rinse with PBS, and add 250µl PBS.
- Assess propidium iodide labelling of the nuclei by epifluorescence microscopy.
- After the 2-h membrane resealing step described in step 4, remove the culture medium, rinse with PBS, and add 250µl PBS containing 0.1µg/ml propidium iodide; incubate for 10 min.
- Assess propidium iodide uptake, or lack thereof, in the resealed cells as in step 6.
E. Reprogramming Reaction
1. Extract Preparation
During SLO treatment, the extract should be prepared for reprogramming.
- Prepare the ATP-regenerating system: mix on ice ATP:GTP:creatine kinase:phosphocreatine in a 1:1:1:1 ratio from each separate stock (described in Section II,A) and keep on ice.
- Add 5µl of the ATP-regenerating system mix to 100 µl of extract on ice.
- Add 4 µl of the 25 mM NTP mix (see Section II,A) to 100 µl of extract on ice.
- Vortex briefly and replace the extract on ice.
2. Reprogramming Reaction
- Remove SLO from the cells by careful aspiration.
- Quickly add PBS to prevent drying.
- Immediately transfer each coverslip into a new dry well of a 24-well plate and carefully lay 65µl of extract (prepared as described in Section III, B,3) onto each coverslip. Be careful that cells do not dry out upon transfer of the coverslip(s) to the new wells and prior to addition of the extract. It is important that the coverslip be covered by the extract during the entire incubation time. Should the extract spread out of the coverslip, transfer the coverslip into a new well and pipette the extract back onto the cells.
- Incubate at 37°C for 1h in regular atmosphere. Note that reprogramming has also proven to be successful upon incubation in a 5% CO2 incubator at 37°C.
E Resealing Reprogrammed Cells
- At the end of incubation, directly add to each well 1.5 ml of preheated (37°C) complete RPMI 1640 containing 2mM Ca2+ added from the 1M stock (see Section II,A). Do not remove the extract before adding the Ca2+-containing medium.
- Incubate for 2h in a 5% CO2 incubator at 37°C.
- Remove the Ca2+-containing medium by gentle aspiration and replace with 250 µl of complete RPMI 1640 (Jurkat TAg cell culture medium).
- Place the cells back into the 5% CO2 incubator and culture until reprogramming assessments are performed.
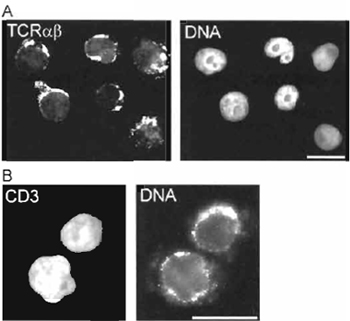 |
| FIGURE 3 Immunofluorescence detection of (A) T-cell receptor (TCR) α and β chain expression and (B) CD3 expression on the surface of 293T cells reprogrammed in a Jurkat TAg cell extract. DNA is labelled with 0.1 µg/ml propidium iodide. Analysis was performed ~14 days after the reprogramming reaction. Bars: 20µm. |
Various assessments of nuclear and cell reprogramming can be performed, depending on the purpose of the experiment. Using the method described here, we have reported changes in the gene expression profile of the reprogrammed 293T cells using cDNA macroarrays from R&D Systems (Abington, UK) (Håkelien et al., 2002). Expression of new proteins can also be monitored at regular intervals after the reprogramming reaction by immunofluorescence or flow cytometry using standard protocols. We have shown the expression of several antigens on the surface of the reprogrammed cells, which are specific for hematopoietic cells. These include CD3, CD4, CD8, CD45, and components of the T-cell receptor (TCR) complex (Håkelien et al., 2002). A variety of functional assays can also be carried out, such as cytokine secretion in response to stimulation of the T-cell receptor/CD3 complex in the reprogrammed cells, or expression of additional cytokine receptors on the cell surface (Håkelien et al., 2002). Fig. 3 illustrates the expression of the TCR α and β chains and of CD3 molecules on the surface of 293T cells reprogrammed in a Jurkat TAg cell extract.
IV. COMMENTS
- Commercially available SLO batches vary greatly in activity. Thus, it is recommended to test a range of SLO concentration on the cell type to be reprogrammed prior to initiating reprogramming reactions. The efficiency of SLO-mediated permeabilisation also varies for various cell types.
- pH of the extract. We usually observe a drop of 1-1.5 pH unit upon extract preparation, which explains the pH 8.2 of the CLB. Notably, raising the pH of CLB to 8.7 with a HEPES buffer does not increase the pH of the final extract. Other buffers with greater buffering capacity have not been tested.
- The method described here can be used easily with either purified cell nuclei (Landsverk et al., 2002) or permeabilised cells (Hakelien et al., 2002). Procedures for purifying intact (membrane-enclosed) nuclei from interphase cultured cells have been reported earlier (Collas et al., 1999; Landsverk et al., 2002; Martins et al., 2000; Steen et al., 2000).
V. PITFALLS
- It is currently difficult to objectively assess the extent of sonication of Jurkat TAg cells or any other cell type. It is important to sonicate until all cells and nuclei are completely lysed. Whether extended sonication after cell lysis is complete is detrimental or beneficial is at present unknown.
- Variability in batches of reprogramming extracts is seen, even among extracts that have been rated as "nontoxic" in the toxicity assay described in Section III,B,4. Variability is evident by the absence of markers of cell reprogramming approximately I week after the reprogramming reaction.
- Currently, the duration of expression of a reprogrammed phenotype is limited to at least 2 months for 293T cells reprogramming in Jurkat TAg extract (Håkelien et al., 2002). The reprogrammed phenotype may also last for shorter periods depending on the marker analyzed.
Acknowledgments
Our work was supported by Nucleotech, LLC and grants from the Research Council of Norway, the Norwegian Cancer Society, and the Human Frontiers Science Program.
References
Blau, H. M., and Blakely, B. T. (1999). Plasticity of cell fate: Insights from heterokaryons. Semin. Cell D 10, 267-272.
Cibelli, J. B., Stice, S. U, Golueke, P. J., Kane, J. J., Jerry, J., Blackwell, C., Ponce, D. L. E, and Robl, J. M. (1998). Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science 280, 1256-1258.
Collas, P., Le Guellec, K., and Tasken, K. (1999). The A-kinase anchoring protein, AKAP95, is a multivalent protein with a key role in chromatin condensation at mitosis. J. Cell Biol. 147, 1167-1180.
Gurdon, J. B., Laskey, R. A., De Robertis, E. M., and Partington, G. A. (1979). Reprogramming of transplanted nuclei in amphibia. Int. Rev. Cytol. Suppl. 161-178.
H~kelien, A. M., Landsverk, H. B., Robl, J. M., Skålhegg, B. S., and Collas, P. (2002). Reprogramming fibroblasts to express T-cell functions using cell extracts. Nature Biotechnol. 20, 460-466.
Landsverk, H. B., Håkelien, A. M., Kfintziger, T., Robl, J. M., Skålhegg, B. S., and Collas, P. (2002). Reprogrammed gene expression in a somatic cell-free extract. EMBO Rep. 3, 384-389.
Martins, S. B., Eide, T., Steen, R. L., Jahnsen, T., Skålhegg, B. S., and Collas, P. (2000). HA95 is a protein of the chromatin and nuclear matrix regulating nuclear envelope dynamics. J. Cell Sci. 113, 3703-3713.
Morrison, S. J. (2001). Stem cell potential: Can anything make anything? Curr. Biol. 11, R7-R9.
Munsie, M. J., Michalska, A. E., O'Brien, C. M., Trounson, A. O., Pera, M. E, and Mountford, P. S. (2000). Isolation of pluripotent embryonic stem cells from reprogrammed adult mouse somatic cell nuclei. Curr. Biol. 10, 989-992.
Steen, R. L., Martins, S. B., Tasken, K., and Collas, P. (2000). Recruitment of protein phosphatase 1 to the nuclear envelope by Akinase anchoring protein AKAP149 is a prerequisite for nuclear lamina assembly. J. Cell Biol. 150, 1251-1262.
Tada, M., Takahama, Y., Abe, K., Nakastuji, N., and Tada, T. (2001). Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr. Biol. 11, 1553-1558.
Walev, I., Hombach, M., Bobkiewicz, W., Fenske, D., Bhakdi, S., and Husmann, M. (2002). Resealing of large transmembrane pores produced by streptolysin O in nucleated cells is accompanied by NF-kappaB activation and downstream events. FASEB J. 16, 237-239.
Western, p. S., and Surani, M. A. (2002). Nuclear reprogramming. Alchemy or analysis? Nature Biotechnol. 20, 445-446.
Wilmut, I., Schnieke, A. E., McWhir, J., Kind, A. J., and Campbell, K. H. S. (1997). Viable offspring derived from fetal and adult mammalian cells. Nature 385, 810-813.
Support our developers




