Tertiary Structure
A. Protein Folding RulesExamination of a large number of protein structures has yielded a few common rules about the folds that proteins can adopt as listed in the following. The theoretical basis for these features is not well understood, but most appear to result from the chirality of the amino acids, entropic considerations, and the necessity to establish a hydrophobic core.
- Secondary structural elements that are close in the
sequence of a protein are often adjacent in the folded
protein. It is less common to find secondary structural
elements that are far apart in the sequence and close
together in the structure. The exception to this arises
where an auxiliary domain has clearly been inserted
into a loop in a protein. This is probably the most
entropically favorable way to arrange secondary
structural elements within a folded protein.
- Adjacent parallel β-strands are almost exclusively
connected by right-handed crossovers (Fig. 9). It is
believed that this feature arises from the chirality of
the amino acids that leads to a net right-handed twist
in the polypeptide chain.
- There are no topological knots in proteins.
- Proteins always contain more than one layer of
secondary structural elements. This rule arises
because proteins always contain a hydrophobic core
formed by the association of hydrophobic side chains.
- α-Helices and β-sheets typically associate in discrete layers of the same type of secondary structural elements. This feature is the consequence of the necessity to fulfill the hydrogen bonding requirements of the polypeptide chain and packing considerations. Typically the interior of a protein does not contain any holes larger than a water molecule. Because α-helices and β-strands differ greatly in their cross-sectional diameters, inclusion of these in the same layer would result in a poorly packed protein interior. In addition a mixture of α-helices and β-strands in the same layer would not fulfill the hydrogen bonding potential of the β-strand.
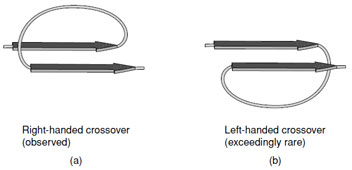 |
| FIGURE 9 A right-handed cross-over connection joining two parallel β-strands. Note the right-handed twist of the β-strands when viewed along the strand axis. |
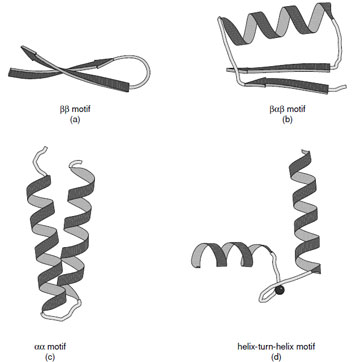 |
| FIGURE 10 Ribbon representations of the (a) ββ, (b) βαβ, (c) αα motif, and (d) helix-turn-helix motif. Various forms of the αα motifs are found depending on the manner in which the α-helices associate. (c) Shows the alignment of two helices joined by a short connection. (d) Shows the helix–turn–helix motif associated with calcium binding proteins. |
B. Folding Motifs
Many protein structures are dominated by a few simple folding motifs. These represent thermodynamically favorable arrangements of secondary structural elements. These include the ββ, βαβ, and αα motifs as illustrated in Fig. 10.
The ββand βαβmotifs are commonly used to connect antiparallel and parallel β-strands, respectively. The ββmotif is frequently connected by a hairpin turn, which provides a compact way of changing the direction of the polypeptide chain. In the same way, the βαβmotif provides a compact module where the width of the α-helix is similar to that of the combined width of the two β-strands. It also provides a hydrophobic core. The dimensions of the βαβmotif explain why large parallel sheets that are built with this motif always have α-helices on both sides since there is insufficient space on one side of a sheet to accommodate all of the connecting helices.
A variety of αα motifs are found in proteins depending on whether the α-helices are in contact with each other after the connecting loop. In cases where the α-helices are in contact they are typically inclined at an angle of either 20 or 50° reflecting the optimal ways to interdigitate side chains at their intersection. Both types of interaction are abundant in proteins and give rise to parallel or crossed helical bundles. There are also many important examples of αα motifs where the connections between the two helices are longer to create a ligand binding site. Important examples of this type of motif are the helix–turn–helix motifs found in calcium binding proteins and DNA binding proteins.
C. Domains
The tertiary structure of a protein describes the manner in which the secondary structural elements are arranged in three dimensions to create a stable molecular entity. In many cases it is convenient to describe a protein in terms of regions of the polypeptide chain that might fold autonomously. These regions are called domains and much of the discussion of tertiary structure centers on classification of these units of protein structure.
Domains in proteins take on many forms. On some occasions it is clear that domains are connected by flexible hinge regions and that the domains could be expressed independently. In other cases the domains are built from apparently distant segments of the protein sequence such that it would be difficult to express those domains without rearrangement of the DNA. This illustrates an important difference in the use of “domain” in structural and molecular biology, since in the latter the term usually indicates a linear section of DNA that appears to influence a biological property where as in structural biology it represents an three-dimensional entity.
D. Protein Folds
Structural studies on proteins have uncovered a very wide variety of protein folds. At this time the upper limit of the number of unique ways in which proteins can fold is unknown; however, genomic sequencing has provided a limit for the maximum number of folds that might be needed for the life of an organism by providing an upper limit to the number of proteins in the genome. Fortunately, the number of unique folds is likely to be considerably less than the total number of proteins since many proteins of dissimilar function have been found to contain the same fold.
The assortment of protein folds observed thus far, at first glance, appears bewilderingly complex. Careful analysis of the common structural and topological features of these structures has lead to a classification of protein folds according to the content and arrangement of the α-helices and β-strands. In turn, this has provided insight into the common underlying principles of protein structure. Several important databases exist of protein structures and tertiary structure classification. These include the RSCB (//www.rcsb.org/pdb/), CATH classification (//www.biochem.ucl.ac.uk/bsm/cath/), and SCOP (//scop.mrc-lmb.cam.ac.uk/scop/) structural databases. The first includes all of the coordinates for structures that have been made publicly available. The second two databases contain structural classifications for all the protein deposited in the RSCB. Both of these systems initially classify proteins into five major groups: all α, all β, α/β (where these secondary structural elements alternate through the fold), α +β (where the sections containing these secondary structural elements are segregated), and small proteins that are stabilized by metal ligands or disulfide bonds. Additional classifications have been added to incorporate multidomain proteins, membrane proteins, and peptides. Representative members of these families are discussed in the following.
E. All α-Proteins
Proteins that fall into this class typically consist of predominately α-helices (>60%), but may contain a small amount of β-sheet at their periphery (See CATH Classification). Historically the first two protein structures determined, myoglobin and hemoglobin, belonged to the all-α class.
These are representative members of the crossed α-helical bundle motif and demonstrate one of the effective ways of packing helices into a protein core (Fig. 11a). The fourhelix bundle illustrated in Fig. 11b shows the second way in which helices associate. When the connecting loops are short the packing leads to an antiparallel arrangement of helices; however, mixtures of parallel and antiparallel helices are also observed in folds that contain longer intervening sequences.
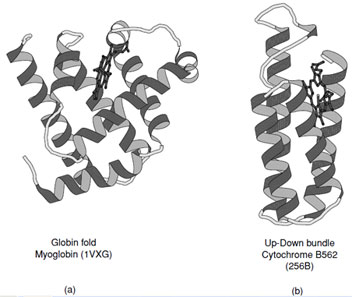 |
| FIGURE 11 Ribbon representations of (a) myoglobin and (b) cytochrome B562. These are representative examples of all α-proteins which exhibit the two major ways of packing α-helices in proteins. |
F. All β-Proteins
There are a large number of diverse protein folds that fall into the all-βclass, but they all contain ∼50% β-strand with only a very small amount of α-helix. Several representative examples of these are shown in Fig. 12. In structures composed primarily of β-strands there is always more than one layer which is necessary to establish a hydrophobic core. This means that at the edge of the sheets there must be some form of compensation for the disruption of the hydrogen bonding pattern. Most of the folds in this class are formed from antiparallel arrangements of β-strands.
A large number of the all-βstructures arrange their strands to form barrel-like or sandwich structure. The simplest of these arrangements is the up-and-down barrel or “clam motif” found in the retinol binding superfamily of proteins (Fig. 12a) where each strand adds to the next in an antiparallel manner until the barrel is complete. In this group of proteins the interior of the barrel provides a binding site for hydrophobic ligands.
Asubstantial number of all-βproteins are built from antiparallel strands in which adjacent strands are not directly connected. Many of these contain a topological feature known as a Greek Key in which the first strand connects across the top of the barrel or sandwich to the fourth strand which then returns via two hairpin connections to the strand adjacent to the first. This feature contains a handedness that is only observed in one sense as shown in Fig. 13a. Connection of two of these features gives rise to an eight stranded β-barrel; however, Greek Key motifs are utilized in many ways to form closed structures. An alternative way of forming a closed barrel is found in proteins that exhibit a “jelly roll” topology (Fig. 13b). This is an abundant motif that is commonly found in virus capsid proteins.
The inclusion of a few sections of random coil or the occasional α-helix into an all-βprotein allows for the generation of some remarkable motifs as shown in the β-propellors and β-helical folds. These folds illustrate the versatility of the β-strand when the hydrogen bonding potential of the polypeptide chain is fulfilled.
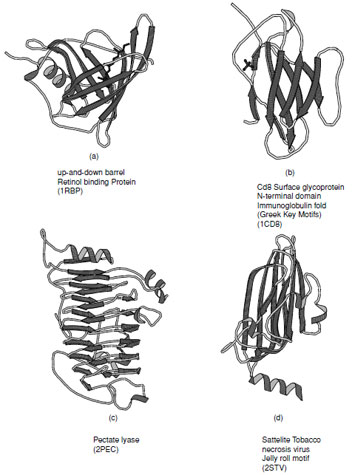 |
| FIGURE 12 Ribbon representations of typical all-βproteins. (a) Retinol binding protein, (b) immunoglobulin fold as seen in the Cd8 Surface glycoprotein N-terminal domain, (c) pectate lyase, and (d) viral coat protein found in satellite tobacco necrosis virus. |
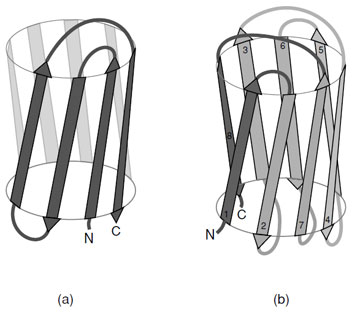 |
| FIGURE 13 Schematic representations of (a) the “Greek Key” and (b) the “jelly roll” topologies commonly found in “all-β” proteins. These topological connections of strands have a handedness where only these arrangements are observed. |
G. α/β Proteins
The α/β class of proteins contains many of the folds that incorporate parallel β-sheets. These folds exist in two major subclasses: the first contains a closed circular β-sheet surrounded by α-helices which forms a barrel; the second is based on an open sheet typically surrounded on both sides by α-helices (Fig. 14). Both of these arrangements are abundant in biosynthetic enzymes.
1. TIM Barrel
Triosephosphate isomerase was the first enzyme shown to contain an (α/β)8 barrel and thus established this motif as the TIM barrel. This fold consists of eight parallel β-strands connected by right-handed helical crossovers and is one of the most common folds found in enzymes. The TIM barrel typically contains approximately 200 amino acid residues. Contrary to the appearance of the ribbon drawing (Fig. 14a), the interior of the barrel is closely packed by the side chains protruding from the β-strands. The strands are inclined at an angle of approximately 30◦ to barrel axis, which is necessary to allow efficient packing of the interior. The necessity to form a closely packed interior explains why these barrels are almost always formed from eight strands. There are several variations on the TIM barrel that include the addition and subtraction of β-strands as well as the introduction of antiparallel β-strands as observed in enolase. These variations attest to the versatility of this fold.
The active sites of triosphosphate isomerase and all other enzymes that contain to this fold are located at the C-terminal end of the β-strands. Typically the catalytic residues reside at the end of the strands and are distributed around the barrel. The loops that connect the strands to the α-helices normally provide the components necessary for substrate specificity. The length of these connecting loops is enormously variable, whereas the length of the β-strands are similar in all enzymes.
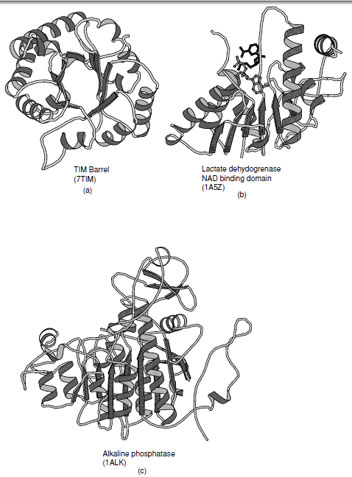 |
| FIGURE 14 Ribbon representations of α/βproteins. (a) Triosephosphate isomerase, (b) dinucleotide binding domain of lactate dehydrogenase (c) alkaline phosphatase which is an example of a complex member of the α/βclass of folds. |
2. Open β-Sheets
The second class of proteins in the α/βfamily of folds contains a large open sheet formed from mostly parallel β-strands with helices on both sides. In contrast to the TIM barrel there are fewer limitations on the number of strands within the sheet and may vary from 4 to 10. The first example of this type of fold was seen in lactate dehydrogenase which contains a motif that is widely observed in dinucleotide binding proteins (this motif is often referred to as the Rossmann fold) and was the first example of a domain superfamily (Fig. 14b). The observation of a common fold in the dehydrogenases by Rossmann and coworkers started the entire field of structural comparison and study of structural evolution.
All of the connections between β-strands are formed by right-handed crossovers. As a consequence, the strand order within the sheet must reverse in order to place helices on both sides of the sheet (Note: the consecutive strand order in the (α/β)8 barrel places the α-helices on one side of the sheet). In the classical Rossmann fold, which contains six β-strands, the N-terminal strand in the fold is located adjacent to the center of the motif. The first two α-helices lie on one side of the sheet as the first three strands are added. Thereafter the chain returns to the center of the sheet and adds the next three strands with the reverse strand order such that the subsequent helices are added on the opposite side of the sheet.
There are many varieties of open sheet α/βproteins which include differing numbers of strands, connections between strands that are not adjacent and incorporation of antiparallel strands. In most cases the ligand binding sites arelocatedat theC-terminal ends of the β-strands andlieat the crevice at the edge of the sheet where the strand order is reversed. The loops that connect the strands to the helices typically provide the residues necessary for specificity. The size of the connecting loops are enormously variable in α/βproteins.
H. α+β Proteins
The α+β class of proteins is highly variable, indeed over a hundred distinct folds have been observed in this group. Members of this class typically contain one or more β-sheets which have a bias toward antiparallel connections. As such the α-helical and β-sheet regions tend to be segregated along their sequences. Several examples of proteins that fall in this class are shown in Fig. 15. In the simplest cases the helices lie on one side of the sheet which maybecomparativelyflat or steeplycurvedas inubiquitin. These are known as αβfolds. In more complex folds multiple layers of sheet and additional layers of helices have been observed to give rise to αββ (as in ribonuclease) and αββα folds (as in glutamine phosphribosyl pyrophosphate amidotransferase N-terminal domain).
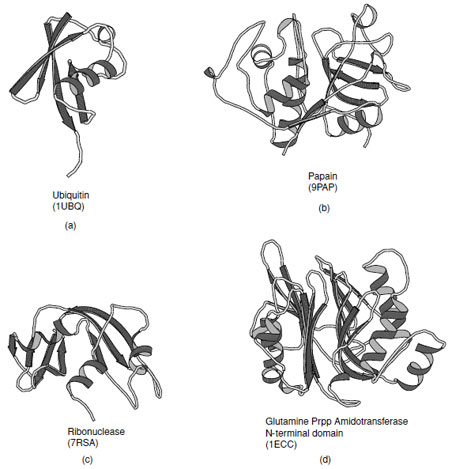 |
| FIGURE 15 Examples of α+βproteins. (a) Ubiquitin, (b) Papain consists of one alpha-helix and four strands of antiparallel beta-sheet, (c) ribonuclease A, and (d) N-terminal domain of Escherichia coli glutamine phosphoribosylpyrophosphate (Prpp) amidotransferase that contains a four structural layers; αββα. |
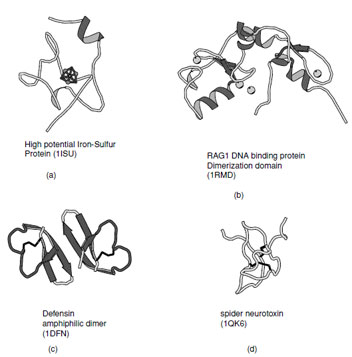 |
| FIGURE 16 Ribbon representation of irregular structures: (a) High potential iron–sulfur protein coordinated to a 4Fe–4S cluster, (b) RAG1 DNA binding protein which contains representative examples of Zn–finger domains, (c) Defensin as example of a membrane toxin stabilized by disulfide bonds, and (d) Chinese bird spider neurotoxin which contains a cystine knot. |
I. Small Proteins, Unusual Folds
There are a substantial number of small proteins that defy classification into one of the groups listed above. Some of these have limited regular secondary structure whereas others are stabilized by metal ligands, cofactors, or disulfide bonds. Examples of thesefolds includethezinc–finger DNA binding motifs, many small iron–sulfur proteins, toxins and protein-inhibitors (Fig. 16).
Support our developers




