Modified Benzyltetrahydroisoquinoline Alkaloids
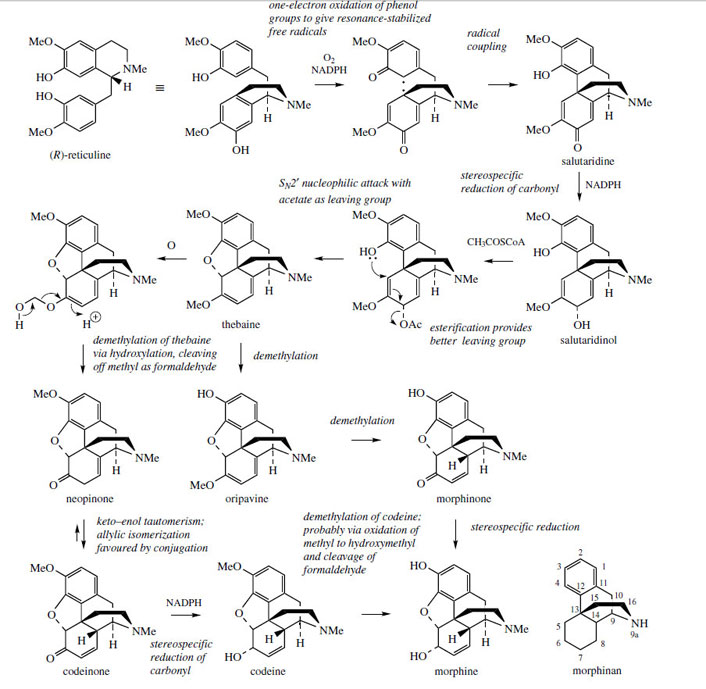
The concept of phenolic oxidative coupling is a crucial theme in modifying the basic benzyltetrahydroisoquinoline skeleton to many other types of alkaloid. Tetrandrine (Figure 46) and tubocurarine (Figure 47) represent coupling of two benzyltetrahydroisoquinoline molecules by ether bridges, but this form of coupling is perhaps less frequent than that involving carbon–carbon bonding between aromatic rings. The principal opium alkaloids morphine, codeine, and thebaine (Figure 50) are derived by this type of coupling, though the subsequent reduction of one aromatic ring to some extent disguises their benzyltetrahydroisoquinoline origins. (R)-Reticuline (Figure 45) is firmly established as the precursor of these morphinan alkaloids. |
 |
| Figure 50 |
(R)-Reticuline, rewritten as in Figure 50, is the substrate for one-electron oxidations via the phenol group in each ring, giving the diradical. Coupling ortho to the phenol group in the tetrahydroisoquinoline, and para to the phenol in the benzyl substituent, then yields the dienone salutaridine, found as a minor alkaloid constituent in the opium poppy Papaver somniferum (Papaveraceae). Only the original benzyl aromatic ring can be restored to aromaticity, since the tetrahydroisoquinoline fragment is coupled para to the phenol function, a position which is already substituted. The alkaloid thebaine is obtained by way of salutaridinol, formed from salutaridine by stereospecific reduction of the carbonyl group. Ring closure to form the ether linkage in thebaine would be the result of nucleophilic attack of the phenol group on to the dienol system and subsequent displacement of the hydroxyl. This cyclization step can be demonstrated chemically by treatment of salutaridinol with acid. In vivo, however, an additional reaction is used to improve the nature of the leaving group, and this is achieved by
acetylation with acetyl-CoA. The cyclization then occurs readily, and without any enzyme participation. Subsequent reactions involve conversion of thebaine into morphine by way of codeine, a process which modifies the oxidation state of the diene ring, but most significantly removes two O-methyl groups. One is present as an enol ether, removal generating neopinone, which gives codeinone and then codeine by allylic isomerization and reduction respectively. The last step, demethylation of the phenol ether codeine to the phenol morphine, is the type of reaction only achievable in the laboratory by the use of powerful and reactive demethylating agents, e.g. HBr or BBr3. Because of the other functional groups present, chemical conversion of codeine into morphine is not usually a satisfactory process. However, the enzymemediated conversion in P. somniferum proceeds smoothly and efficiently. The enzymic demethylations of both the enol ether and the phenol ether probably involve initial hydroxylation followed by loss of the methyl groups as formaldehyde (Figure 50).
The involvement of these O-demethylation reactions is rather unusual; secondary metabolic pathways tend to increase the complexity of the product by adding methyls rather than removing them. In this pathway, it is convenient to view the methyl groups in reticuline as protecting groups, which reduce the possible coupling modes available during the oxidative coupling process, and these groups are then removed towards the endof the synthetic sequence. There is also some evidence that the later stages of the pathway in Figure 50 are modified in some strains of opium poppy. In such strains, thebaine is converted by way of oripavine and morphinone, this pathway removing the phenolic O-methyl before that of the enol ether, i.e. carrying out the same steps but in a different order. The enzymic transformation of thebaine into morphine, and the conversion of (R)-reticuline into salutaridinol have also been observed in mammalian tissues, giving strong evidence that the trace amounts of morphine and related alkaloids which can sometimes be found in mammals are actually of endogenous origin rather than dietary.
Opium
Opium is the air-dried milky exudate, or latex, obtained by incising the unripe capsules of the opium poppy Papaver somniferum (Papaveraceae). The plant is an annual herb with large solitary flowers, of white, pink, or dull red-purple colour. For opium production, the ripening capsules, which are just changing colour from blue-green to yellow, are carefully incised with a knife to open the latex tubes, but not to cut through to the interior of the capsule. These latex tubes open into one another, so it is not necessary to incise them all. Cuts are made transversely or longitudinally according to custom. The initially white milky latex quickly oozes out, but rapidly turns brown and coagulates. This material, the raw opium, is then removed early the following morning, being scraped off and moulded into balls or blocks. Typically, these are wrapped in poppy leaves and shade-dried. The blocks may be dusted with various plant materials to prevent cohering. Fresh opium is pale to dark brown and plastic, but it becomes hard and brittle when stored.
Opium has been known and used for 4000 years or more. In recent times, attempts have been made at governmental and international levels to control the cultivation of the opium poppy, but with only limited success. In endeavours to reduce drug problems involving opium-derived materials, especially heroin, where extremely large profits can be made from smuggling relatively small amounts of opium, much pharmaceutical production has been replaced by the processing of the bulkier 'poppy straw'. The entire plant tops are harvested and dried, then extracted for their alkaloid content in the pharmaceutical industry. Poppy straw now accounts for most of the medicinal opium alkaloid production, but there is still considerable trade in illicit opium. In addition to opium, the opium poppy yields seeds, which are used in baking and are also pressed to give poppy seed oil. The remaining seed cake is used as cattle feed, and it is held that these poppy seed products cover all the growing expenses, with opium providing the profit. Poppy seeds do not contain any significant amounts of alkaloids.
The main producer of medicinal opium is India, whilst poppy straw is cultivated in Turkey, Russia, and Australia. Opium destined for the black market originates from the Golden Triangle (Burma, Laos, and Thailand), the Golden Crescent (Iran, Pakistan, and Afghanistan), and Mexico.
Crude opium has been used since antiquity as an analgesic, sleep-inducer (narcotic), and for the treatment of coughs. It has been formulated in a number of simple preparations for general use, though these are now uncommon. Laudanum, or opium tincture, was once a standard analgesic and narcotic mixture. Paregoric, or camphorated opium tincture, was used in the treatment of severe diarrhoea and dysentery, but is still an ingredient in the cough and cold preparation Gee's linctus. In Dover's powder, powdered opium was combined with powdered ipecacuanha to give a popular sedative and diaphoretic (promotes perspiration) to take at the onset of colds and influenza. Opium has traditionally been smoked for pleasure, but habitual use develops a craving for the drug followed by addiction. An unpleasant abstinence syndrome is experienced if the drug is withdrawn.
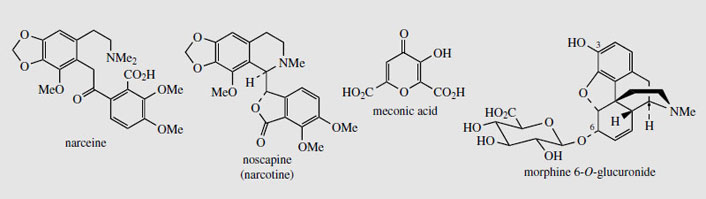
In modern medicine, only the purified opium alkaloids and their derivatives are commonly employed. Indeed, the analgesic preparation 'papaveretum ', which once contained the hydrochlorides of total opium alkaloids, is now formulated from selected purified alkaloids, in the proportions likely to be found in opium. Although the ripe poppy capsule can contain up to 0.5% total alkaloids, opium represents a much concentrated form and up to 25% of its mass is composed of alkaloids. Of the many (>40) alkaloids identified, some six represent almost all of the total alkaloid content. Actual amounts vary widely, as shown by the following figures: morphine (Figure 50) (4-21%); codeine (Figure 50) (0.8-2.5%); thebaine (Figure 50) (0.5-2.0%); papaverine (Figure 45) (0.5-2.5%); noscapine (narcotine) (Figure 51) (4-8%); narceine (Figure 51; Figure 63) (0.1-2%). A typical commercial sample of opium would probably have a morphine content of about 12%. Powdered opium is standardized to contain 10% of anhydrous morphine, usually by dilution with an approved diluent, e.g. lactose or cocoa husk powder. The alkaloids are largely combined in salt form with meconic acid (Figure 51), opium containing some 3-5% of this material. Meconic acid is invariably found in opium, but, apart from its presence in other Papaverspecies, has not been detected elsewhere. It gives a deep red-coloured complex with ferric chloride, and this has thus been used as a rapid and reasonably specific test for opium. In the past, the urine of suspected opium smokers could also be tested in this way.Of the main opium alkaloids, only morphine and narceine display acidic properties, as well as the basic properties due to the tertiary amine. Narceine has a carboxylic acid function, whilstmorphine is acidic due to its phenolic hydroxyl. This acidity can be exploited for the preferential extraction of these alkaloids (principally morphine) from an organic solvent by partitioning with aqueous base.
 |
| Figure 61 |
Morphine (Figure 50) is a powerful analgesic and narcotic, and remains one of the most valuable analgesics for relief of severe pain. It also induces a state of euphoria and mental detachment, together with nausea, vomiting, constipation, tolerance, and addiction. Regular users experience withdrawal symptoms, including agitation, severe abdominal cramps, diarrhoea, nausea, and vomiting, which may last for 10-14 days unless a further dose of morphine is taken. This leads to physical dependence, which is difficult to overcome, so that the major current use of morphine is thus in the relief of terminal pain. Although orally active, it is usually injected to obtain rapid relief of acute pain. The side-effect of constipation is utilized in some anti-diarrhoea preparations, e.g. kaolin and morphine. Morphine is metabolized in the body to glucuronides, which are readily excreted. Whilst morphine 3-O-glucuronide is antagonistic to the analgesic effects of morphine, morphine 6-O-glucuronide (Figure 51) is actually a more effective and longer lasting analgesic than morphine, with fewer side-effects.
Codeine (Figure 50) is the 3-O-methyl ether of morphine, and is the most widely used of the opium alkaloids. Because of the relatively small amounts found in opium, most of the material prescribed is manufactured by semi-synthesis from morphine. Its action is dependent on partial demethylation in the liver to produce morphine, so it produces morphinelike analgesic effects, but little if any euphoria. As an analgesic, codeine has about one-tenth the potency of morphine. Codeine is almost always taken orally and is a component of many compound analgesic preparations. Codeine is a relatively safe non-addictive medium analgesic, but is still too constipating for long-term use. Codeine also has valuable antitussive action, helping to relieve and prevent coughing. It effectively depresses the cough centre, raising the threshold for sensory cough impulses.
Thebaine (Figure 50) differs structurally from morphine/codeine mainly by its possession of a conjugated diene ring system. It is almost devoid of analgesic activity, but may be used as a morphine antagonist. Its main value is as substrate for the semi-synthesis of other drugs.
Papaverine (Figure 45) is a benzylisoquinoline alkaloid, and is structurally very different from the morphine, codeine, thebaine group of alkaloids (morphinans). It has little or no analgesic or hypnotic properties put possesses spasmolytic and vasodilator activity. It has been used in some expectorant preparations, and in the treatment of gastrointestinal spasms, but its efficacy was not substantiated. It is sometimes used as an effective treatment for male impotence, being administered by direct injection to achieve erection of the penis.
Noscapine (Figure 51) is a member of the phthalideisoquinoline alkaloids and provides a further structural variant in the opium alkaloids. Noscapine has good antitussive and cough suppressant activity comparable to that of codeine, but no analgesic or narcotic action. Its original name 'narcotine' was changed to reflect this lack of narcotic action. Despite many years of use as a cough suppressant, the finding that noscapine may have teratogenic properties (i.e. may deform a fetus) has resulted in noscapine preparations being deleted. In recent studies, antitumour activity has been noted from noscapine, which binds to tubulin as do podophyllotoxin and colchicine, thus arresting cells at mitosis. The chemotherapeutic potential of this orally effective agent merits further evaluation.
 |
| Figure 52 |
Papaveretum is a mixture of purified opium alkaloids, as their hydrochlorides, and is now formulated to contain only morphine (85.5%), codeine (7.8%), and papaverine (6.7%). It is used for pain relief during operations.
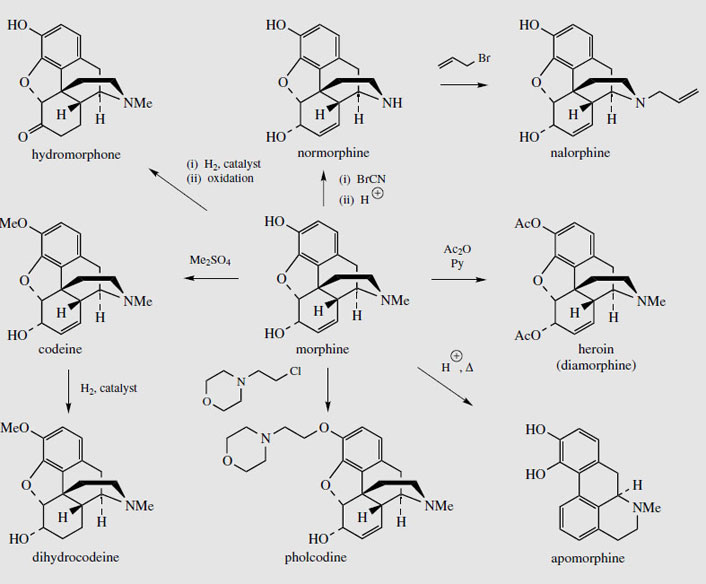
A vast range of semi-synthetic or totally synthetic morphine-like derivatives have been produced. These are collectively referred to as 'opioids'. Many have similar narcotic and pain-relieving properties as morphine, but are less habit forming. Others possess the cough-relieving activity of codeine, but without the analgesic effect. More than 90% of the morphine extracted from opium (or poppy straw) is currently processed to give other derivatives (Figure 52). Most of the codeine is obtained by semi-synthesis from morphine, mono-O-methylation occurring at the acidic phenolic hydroxyl. Similarly, pholcodine (Figure 52), an effective and reliable antitussive, can be obtained by alkylation with N-(chloroethyl)morpholine. Dihydrocodeine (Figure 52) is a reduced form of codeine with similar analgesic properties, the double bond not being essential for activity. In hydromorphone, the double bond of morphine has been reduced, and in addition the 6-hydroxyl has been oxidized to a ketone. This increases the analgesic effects, but also the side-effects. Diamorphine, or heroin (Figure 52) is merely the diacetate of morphine, and is a highly addictive analgesic and hypnotic. The increased lipophilic character results in better transport and absorption, though the active agent is probably the 6-acetate, the 3-acetate group being hydrolysed by esterases in the brain. Heroin was synthesized originally as a cough suppressant, and though most effective in this role has unpleasant addictive properties, with users developing a psychological craving for the drug. It is widely used for terminal care, e.g. cancer sufferers, both as an analgesic and cough suppressant. The euphoria induced by injection of heroin has resulted in much abuse of the drug, and creation of a world-wide major drug problem.
The N-methyl group of morphine can be removed by treatment with cyanogen bromide, then hydrolysis. A variety of N-alkyl derivatives, e.g. N-allyl-normorphine (nalorphine) (Figure 52) may be produced by use of appropriate alkyl bromides. Nalorphine has some analgesic activity, but was also found to counter the effects of morphine, and is thus a mixed agonist-antagonist. It is sometimes used as a narcotic antagonist, but is principally regarded as the forerunner of pure opiate antagonists such as naloxone. Treatment of morphine with hot acid induces a rearrangement process, resulting in a highly modified structural skeleton, a representative of the aporphine group of alkaloids. The product apomorphine (Figure 52) has no analgesic properties, but morphine's side-effects of nausea and vomiting are highly emphasized. Apomorphine is a powerful emetic, and can be injected for emergency treatment of poisoning. This is now regarded as dangerous, but apomorphine is also valuable to control the symptoms of Parkinson's disease, being a stimulator of D1 and D2 dopamine receptors. Apomorphine's structure contains a dihydroxyphenylethylamine (dopamine) fragment, conferring potent dopamine agonist properties to this agent.
 |
| Figure 53 |
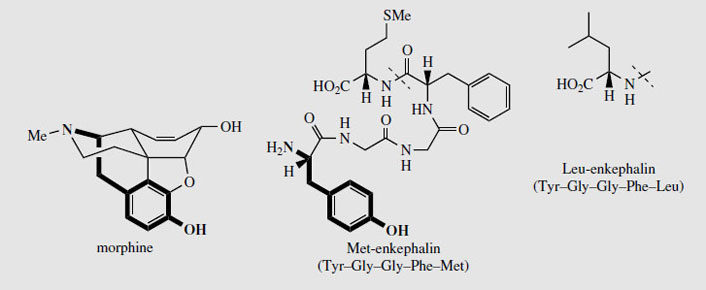
It has been found that a common structural feature required for centrally acting analgesic activity in the opioids is the combination of an aromatic ring, and a piperidine ring which maintain the stereochemistry at the chiral centre as shown in Figure 53. The threedimensional disposition of the nitrogen function to the aromatic ring allows morphine and other analgesics to bind to a pain-reducing receptor in the brain. Several different receptors and groups of receptors are known. The natural agonists include peptides called enkephalins, Met-enkephalin and Leu-enkephalin (Figure 53), produced from a larger peptide endorphin. The terminal tyrosine residue in the enkephalins is mimicked by portions of the morphine structure. The enkephalins themselves are rapidly degraded in the body and are unsuitable for drug use.
 |
| Figure 54 |
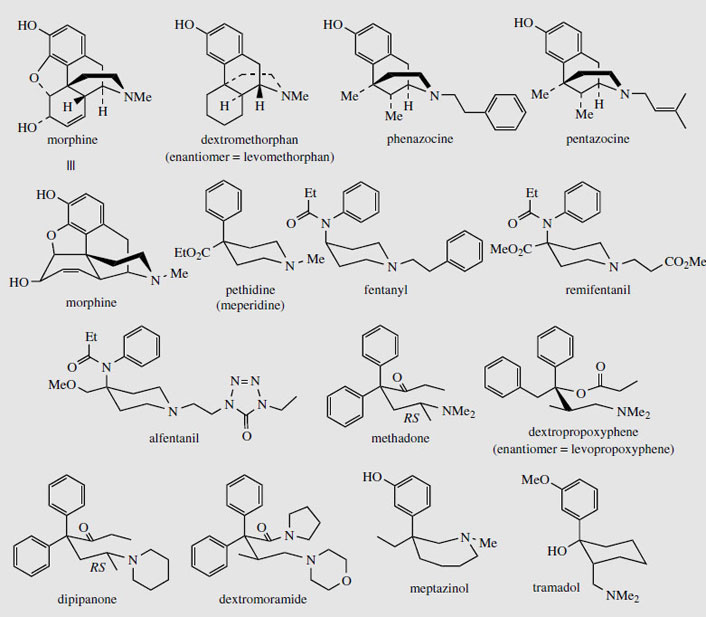
Some totally synthetic opioid drugs modelled on morphine are shown in Figure 54. Removal of the ether bridge and the functionalities in the cyclohexene ring are exemplified in levomethorphan and dextromethorphan. Levomethorphan has analgesic properties, whilst both enantiomers possess the antitussive activity of codeine. In practice, the 'unnatural' isomer dextromethorphan is the preferred drug material, being completely non-addictive and possessing no analgesic activity. Pentazocine and phenazocine are examples of morphinelike structures where the ether bridge has been omitted and the cyclohexene ring has been replaced by simple methyl groups. These drugs are good analgesics and are non-addictive, though pentazocine can induce withdrawal symptoms. Even more drastic simplification of the morphine structure is found in pethidine (meperidine), one of the most widely used synthetic opiates. Only the aromatic ring and the piperidine systems are retained. Pethidine is less potent than morphine, but produces prompt, short-acting analgesia, and is also less constipating than morphine. It can be addictive. Fentanyl has a 4-anilino- rather than a 4-phenyl-piperidine structure, and is 50-100 times more active than morphine due to its high lipophilicity and excellent transport properties. Alfentanil and remifentanil are further variants on the fentanyl structure; all three drugs are rapid-acting and used during operative procedures. The piperidine ring system is no longer present in methadone, though this diphenylpropylamine derivative can be drawn in such a way as to mimic the piperidine ring conformation. Methadone is orally active, has similar activity to morphine, but is less euphorigenic and has a longer duration of action. Although it is as potentially addictive as morphine, the withdrawal symptoms are different and much less severe than with other drugs such as heroin, and methadone is widely used for the treatment and rehabilitation of heroin addicts. However, it only replaces one addiction with another, albeit a less dangerous one. Dextropropoxyphene mimics the piperidine ring in a rather similar manner, but this agent has only low analgesic activity, about half that of codeine, and finds application in combination formulations with aspirin or paracetamol. The enantiomeric levopropoxyphene has antitussive activity, but no analgesic properties. Dipipanone and dextromoramide are structural variants on methadone, and are used for moderate to severe pain; dipipanone is usually administered in combination with an anti-emetic. Meptazinol is structurally unlike the other opiate analgesics in that it contains a seven-membered nitrogen heterocycle. It is an effective analgesic, and produces relatively few side-effects with a low incidence of respiratory depression. Tramadol is a recent drug claimed to produce analgesia by an opioid mechanism and by enhancement of serotoninergic and adrenergic pathways, with few typical opioid side-effects.
 |
| Figure 55 |
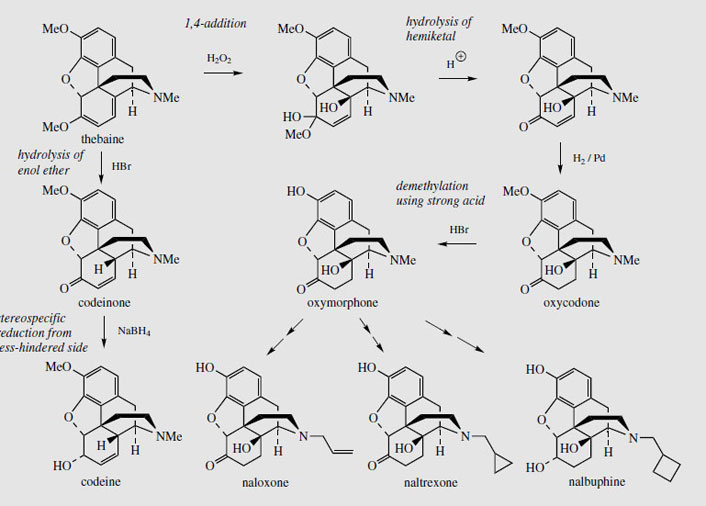
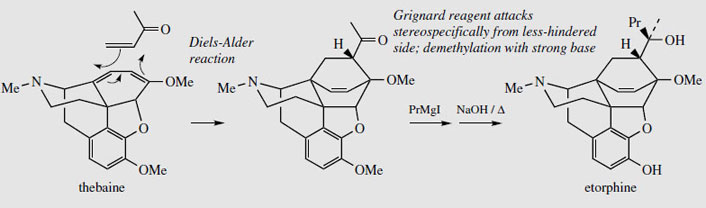
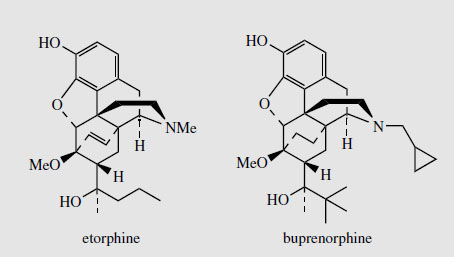
Thebaine, for many years regarded as an unwanted by-product from opium, is now utilized for the semi-synthesis of useful new drugs. On treatment with hydrogen peroxide, the conjugated diene undergoes 1,4-addition, and hydrolysis results in formation of a 4- hydroxy cyclohexenone system (Figure 55). Reduction and demethylation lead respectively to oxycodone and oxymorphone, which are potent analgesics. The conjugated diene system can also be exploited in a Diels-Alder reaction, building on another ring system (Figure 56). Some of these adducts have quite remarkable levels of analgesic activity, but are too powerful for human use. Some, e.g. etorphine (Figure 57), are used in veterinary practice to sedate large animals (elephants, rhinos) by means of tranquillizer darts. Etorphine is some 5000-10 000 times more potent than morphine. Buprenorphine (Figure 57) is an etorphine analogue with an N-cyclopropylmethyl substituent and tert-butyl instead of N-propyl in the side-chain. This material has both opioid agonist and antagonist properties. Mixed agonist-antagonist properties offer scope for producing analgesia whilst negating the effects of other opioids to which a patient may be addicted. Buprenorphine has a long duration of action, and only low dependence potential, but may precipitate withdrawal symptoms in patients dependent on other opioids. It is now being used as an alternative to methadone in the treatment of opioid dependence. Nalbuphine (Figure 55), produced semi-sythetically from thebaine, also displays mixed agonist-antagonist properties, and has similar agonist activity as morphine, but produces less side-effects and has less abuse potential. Naloxone(Figure 55) shows hardly any agonist activity but is a potent antagonist, and is used to treat opiate poisoning, including that in children born to heroin addicts. Naltrexone (Figure 55) also has antagonist activity similar to naloxone. These agents are N-alkyl derivatives related to oxymorphone/oxycodone.
 |
| Figure 56 |
 |
| Figure 57 |
Thebaine may also be transformed very efficiently into codeine in about 75% yield (Figure 55). The two-stage synthesis involves acid-catalysed hydrolysis of the enol ether function to give codeinone (this being the more favoured tautomer of the first-formed β, γ- unsaturated ketone) followed by selective borohydride reduction of the carbonyl. This opens up possibilities for producing codeine (the most widely used of the opium alkaloids) without using morphine. At present, most of the codeine is synthesized by methylation of morphine. The advantage of using thebaine is that the raw material for the pharmaceutical industry could be shifted away from morphine and opium. This might then help in the battle to eliminate illicit morphine production and its subsequent conversion into heroin. Conversion of thebaine into morphine and heroin is much more difficult and low yielding. Thus, there is interest in cultivating Papaver bracteatum rather than P. somniferum. This plant produces mainly thebaine, no morphine, and only faint traces of codeine. Experiments have shown it has the enzymic activity to convert codeinone into codeine, but it appears to lack enzymes which carry out the late demethylation steps in Figure 50. The capsules can produce up to 3% thebaine, but, regrettably, there have been difficulties in making this a commercially viable project, and this idea has not materialized. Other species of Papaver seem to lack the enzyme that reduces salutaridine to salutaridinol (Figure 50) and they thus do not synthesize morphine-like alkaloids. Remarkably, there is now considerable evidence that various animals, including humans and other mammals, are also able to synthesize morphine and related alkaloids in small amounts. These compounds have been detected in various tissues, including brain, liver, spleen, adrenal glands and skin, and endogenous morphine may thus play a role in pain relief, combining its effects with those provided by the enkephalin peptides.
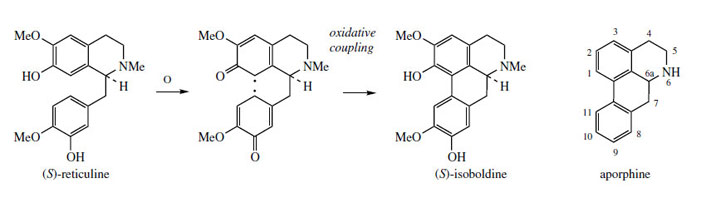
A minor constituent of P. somniferum is the aporphine alkaloid isoboldine (Figure 58). Other species of poppy, e.g. Papaver orientale and P. pseudoorientale, are known to synthesize aporphine alkaloids as principal constituents rather than morphinan structures. (S )-Isoboldine is readily appreciated to be the product of oxidative coupling of (S )-reticuline, coupling ortho to the phenol group in the tetrahydroisoquinoline, and para to the phenol of the benzyl substituent (Figure 58). Some structures, e.g. isothebaine(Figure 59) from P. orientale, are not as easily rationalized. (S )-Orientaline is a precursor of isothebaine (Figure 59). This benzyltetrahydroisoquinoline, with a different methylation pattern to reticuline, is able to participate in oxidative coupling, but inspection of the structures indicates a phenol group is lost in the transformation. The pathway (Figure 59) involves an unexpected rearrangement process, however.
 |
| Figure 58 |
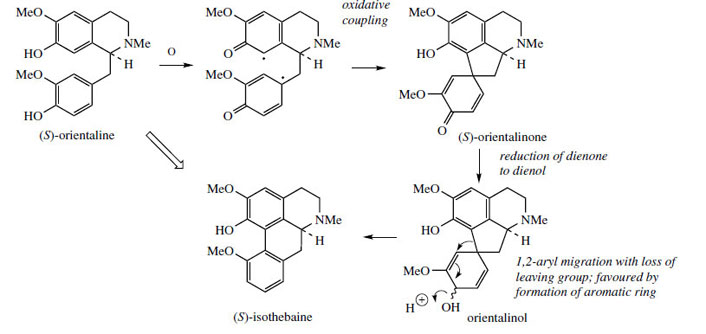
Thus, oxidative coupling ortho–para to the phenol groups gives a dienone orientalinone (compare the structure of salutaridine (Figure 50)). After reduction of the carbonyl group, a rearrangement occurs, restoring aromaticity and expelling the hydroxyl (originally a phenol group) to produce isothebaine. This type of rearrangement, for which good chemical analogies are available, is a feature of many other alkaloid biosynthetic pathways, and occurs because normal keto–enol tautomerism is not possible for rearomatization when coupling involves positions already substituted. The process is fully borne out by experimental evidence, including the subsequent isolation of orientalinone and orientalinol from P. orientale.
 |
| Figure 59 |
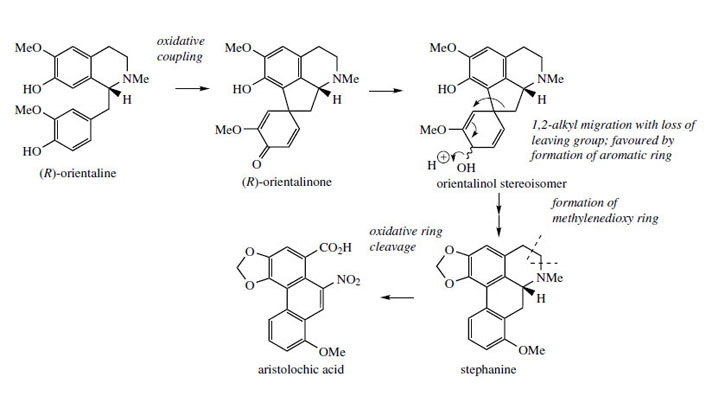
Stephanine (Figure 60) from Stephaniaspecies (Menispermaceae) is analogous to isothebaine and shares a similar pathway, though from (R)-orientaline. The different substitution pattern in stephanine compared to isothebaine is a consequence of the intermediate dienol suffering migration of the alkyl rather than aryl group (Figure 60). Aristolochic acid is a novel modified aporphine containing a nitro group and is produced from stephanine by oxidative Aristolochic acid is present in many species of Aristolochia (Aristolochiaceae) used in traditional medicine, e.g. snake-root A. serpentina. However, because aristolochic acid is now known to be nephrotoxic and to cause acute kidney failure, the use of Aristolochia species in herbal medicines, especially Chinese remedies, has been banned in several countries.
 |
| Figure 60 |
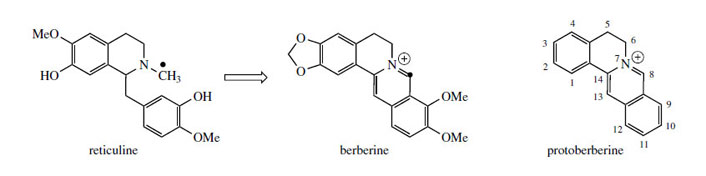
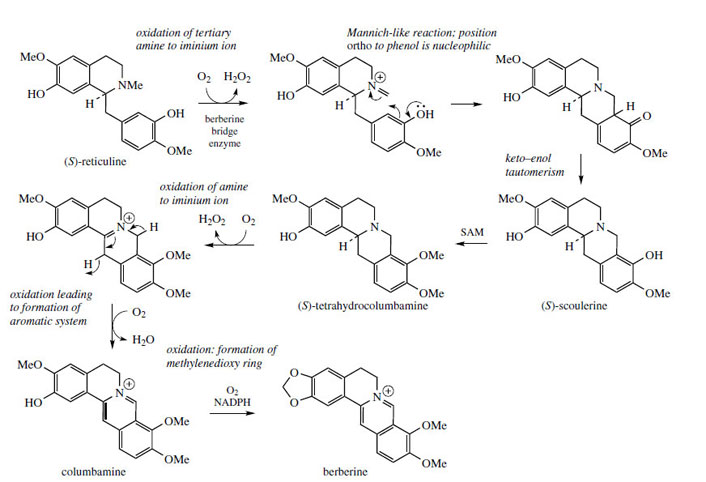
The alkaloid berberine (Figure 61) is found in many members of the Berberidaceae (e.g. Berberis, Mahonia), the Ranunculaceae (e.g. Hydrastis), and other families. Berberine has antiamoebic, antibacterial, and anti-inflammatory properties and plants containing berberine have long been used in traditional medicine. Its tetracyclic skeleton is derived from a benzyltetrahydroisoquinoline system with the incorporation of an extra carbon atom, supplied from S-adenosylmethionine via an N-methyl group (Figure 61). This extra skeletal carbon is known as a ‘berberine bridge’. Formation of the berberine bridge is readily rationalized as an oxidative process in which the N-methyl group is oxidized to an iminium ion, and a cyclization to the aromatic ring occurs by virtue of the phenolic group
(Figure 62).
The oxidative cyclization process is analogous to the formation of a methylenedioxy group, whilst the mechanism of cyclization is exactly the same as that invoked in formation of a tetrahydroisoquinoline ring, i.e. a Mannich-like reaction. The product from the enzymic transformation of (S )-reticuline is the protoberberine alkaloid (S )-scoulerine, the berberine bridge enzyme requiring molecular oxygen as oxidant and releasing H2O2 as by-product (Figure 62). Its role in the cyclization reaction completed, the phenol group in scoulerine is then methylated, and tetrahydrocolumbamine is oxidized further to give the quaternary isoquinoline system in columbamine. This appears to involve two separate oxidation steps, both requiring molecular oxygen, though H2O2 and H2O are produced in the successive processes. The mechanistic sequence through an iminium ion has been suggested to account for these observations. Finally, berberine is produced by transformation of the ortho-methoxyphenol to a methylenedioxy group, via the O2-, NADPH-, and cytochrome P-450- dependent enzyme.
 |
| Figure 61 |
 |
| Figure 62 |
 |
| Figure 63 |
 |
| Figure 64 |
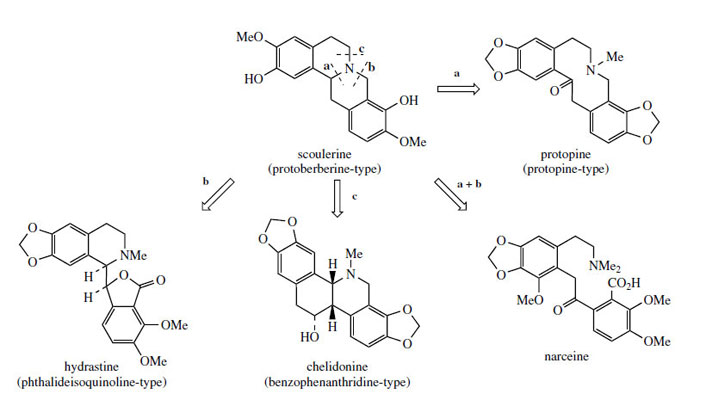
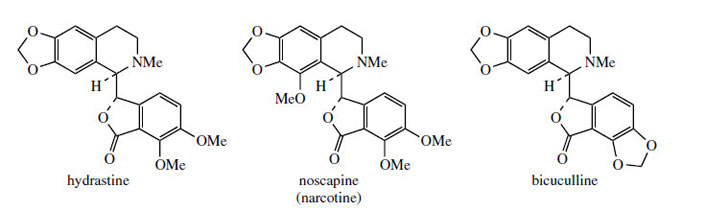
The protoberberine skeleton of scoulerine may be subjected to further modifications, some of which are given in Figure 63. Cleavage of the heterocyclic ring systems adjacent to the nitrogen atom as shown give rise to new skeletal types: protopine, e.g. protopine from Chelidonium majus (Papaveraceae), phthalideisoquinoline, e.g. hydrastine from Hydrastis canadensis(Ranunculaceae), and benzophenanthridine, e.g. chelidonine, also from Chelidonium majus. The non-heterocyclic system seen in the opium alkaloid narceine from Papaver somniferum can be visualized as the result of cleavage of two of these bonds. Some alkaloids of the phthalide type are medicinally important. Noscapine (Figure 64) is one of the opium alkaloids and although it lacks any analgesic activity it is an effective cough suppressant. Hydrastine is beneficial as a traditional remedy in the control of uterine bleeding. Hydrastis also contains berberine, indicating the close biosynthetic relationship of the two types of alkaloid. Bicuculline (Figure 64) from species of Corydalis and Dicentra (Fumariaceae) and its quaternary methiodide have been identified as potent GABA (γ-aminobutyric acid) antagonists and have found widespread application as pharmacological probes for convulsants acting at GABA neuroreceptors.
Support our developers




