Design and Production of Human Immunodeficiency Virus-Derived Vectors
I. INTRODUCTIONLentiviral vectors (LV) can govern the efficient delivery and stable integration of transgenes both in vitro and in vivo, can transduce a wide range of targets, including stem cells, and can be used for generating transgenic animals from several species. Lentiviral vector-mediated gene transfer results in the ubiquitous, tissue-specific, and/or regulated expression of these transgenes, depending on the promoter contained in the vector. Finally, this gene delivery system can mediate the knockdown of endogenous genes by RNA interference via the polymerase III promoterdriven production of small hairpin RNAs.
II. DESIGN
The potential of lentiviral vectors was first revealed in 1996 through the demonstration that they could transduce neurons in vivo (Naldini et al., 1996). Since then, many improvements have been brought to achieve high levels of efficiency and biosafety. The principle, however, remains the same and consists of building replication-defective recombinant chimeric lentiviral particles from three different components: the genomic RNA, the internal structural and enzymatic proteins, and the envelope glycoprotein. A schematic diagram of the evolution of human immunodeficiency virus (HIV)-based vectors is represented in Fig. 1. The genomic RNA contains all cis-acting sequences, whereas packaging plasmids contain all the trans-acting proteins necessary for adequate transcription, packaging, reverse transcription, and integration.
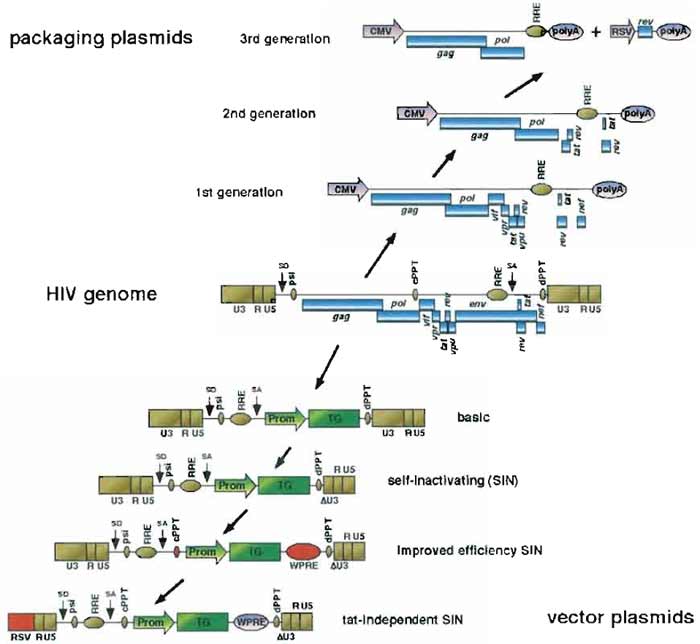 |
| FIGURE 1 Evolution in the design of HIV-l-based LV vectors. HIV-l-based LV vectors are derived from wild-type HIV-1 by dissociation of the trans-acting components (blue boxes, above HIV genome) coding for structural and accessory proteins and the cis-acting sequences required for packaging and reverse transcription of the genomic RNA (golden boxes, below HIV genome). Sequences added between two vector versions are in red. CMV, human cytomegalovirus immediate-early promoter; RRE, rev-responsive element; RSV, Rous sarcoma promoter; polyA, polyadenylation site; U3-R-U5, HIV-1 LTR: SD, major splice donor; psi, HIV-1 packaging signal; cPPT, central polypurine tract; SA, splice acceptor; dPPT, distal (3') polypurine tract; Prom, promoter of the internal expression cassette; TG, transgene of the internal expression cassette; ΔU3, self-inactivating deletion of the U3 part of the HIV-1 LTR; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element. |
A. Envelope
Although various envelope proteins can efficiently pseudotype LV particles (Sandrin et al., 2002), the G protein of the vesicular stomatitis virus (VSV) is the most widely used. The main reasons for this choice are that the VSV envelope (1) allows for high titers achieved in unconcentrated supernatants; (2) provides an extremely wide range for the transduction of target cells (virtually all mammalian cells of any tissue tested so far can be transduced by VSV-G pseudotyped LVs); (3) is very robust, allowing for concentration by ultracentrifugation; and (4) has a good resistance to freeze-thaw cycles.
B. Core and Enzymatic Components ("Packaging System")
The first-generation lentiviral vectors were manufactured using a packaging system that comprised all HIV genes but the envelope (Naldini et al., 1996). In a so-called second-generation system, five of the nine HIV-1 genes were eliminated, leaving the gag and pol reading frames, which encode for the structural and enzymatic components of the virion, respectively, and the tat and rev genes, fulfilling transcriptional and posttranscriptional functions (Zufferey et al., 1997). Sensitive tests have so far failed to detect replicationcompetent recombinants (RCRs) with this system. This good safety record, combined with its high efficiency and ease of use, explains why the second-generation lentiviral vector packaging system is utilized for most experimental purposes. In a third-generation system, geared toward clinical applications, only gag, pol, and rev genes are still present, using a chimeric 5' long terminal repeat (LTR) to ensure transcription in the absence of Tat (see later).
C. Genomic Vector
The genetic information contained in the vector genome is the only one transferred to the target cells. Early genomic vectors were composed of the following components: the 5' LTR, the major splice donor, the packaging signal (encompassing the 5' part of the gag gene), the Rev-responsive element (RRE), the envelope splice acceptor, the internal expression cassette containing the transgene, and the 3' LTR. In the latest generations, several improvements have been introduced. The woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) has been added to increase the overall levels of transcripts in both producer and target cells, hence increasing titers and transgene expression (Zufferey et al., 1999). The central polypurine tract of HIV has also been added back in the central portion of the genome of the transgene RNA (Follenzi et al., 2000; Zennou et al., 2000). This increases titers in some targets. The 3' LTR has been deleted in the U3 region to remove all transcriptionally active sequences, creating the so-called self-inactivating (SIN) LTR (Zufferey et al., 1998). Finally, chimeric 5' LTRs have been constructed in order to render the LV promoter Tat independent. This has been achieved by replacing the U3 region of the 5' LTR with either the CMV enhancer (CCL LTR) or the corresponding Rous sarcoma virus (RSV) U3 sequence (RRL LTR) (Dull et al., 1998). Vectors containing such promoters can be produced at high titers in the absence of the Tat HIV transactivator. However, the Rev dependence of these third-generation LV has been maintained in order to maximize the number of recombination events that would be necessary to generate an RCR (Fig. 2).
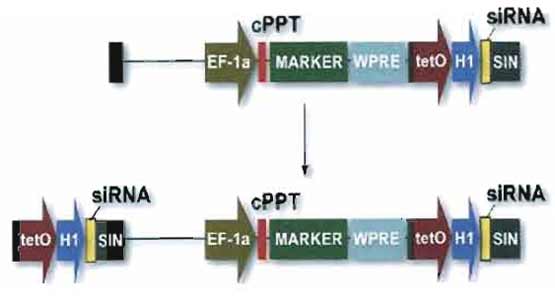 |
| FIGURE 2 Vector for RNA interference. Small hairpin (interfering) RNAs (shRNA or siRNA) are expressed from a polymerase III promoter as described (Brummelkamp et al., 2002). In this example, the expression cassette is placed in the U3 region of the 3' LTR. Because of the modalities of reverse transcription, two copies of the siRNA inducing module will be present in the integrated provirus, facilitating high levels of production. For convenience, a transgene can be placed in the same vector, downstream of an internal promoter. |
III. MATERIALS AND REAGENTS
- 293T and HeLa cell lines (ATCC) (www.atcc.org)
- Chemicals (Sigma-Fluka) (www.sigmaaldrich.com)
- Cell culture media and additives (GIBCO-BRL-Life Technologies) (www.gibcobrl.com) and CellGenix Technologie Transfer GmbH, Germany (www. cellgenix.com)
- Plastics for tissue cultures, flow cytometer (BD Biosciences) (www.bdbiosciences.com)
- DNA purification kits (Qiagen) (wwwl.qiagen.com) and Genomed (www.genomed-dna.com)
- Centrifuges (Sorvall) (www.sorvall.com)
- Filters (Millipore) (www.millipore.com)
- Ultracenrifuge tubes (Beckman) (www.beckman.com)
- Sequence detector ABI7700 plus ABI SDS software for analysis, optical reaction plates and caps for QPCR (Applied Biosystems) (http://www. appliedbiosystems.com)
- QPCR reaction mixes and probes (Eurogentech) (www.eurogentec.com)
- HIV-1 p24 antigen capture assay (AIDS Vaccine Program) (Frederick, MD. email: schadent@mail. ncifcrf.govmhttp://web.ncifcrf.gov)
IV. PRODUCTION
The production of LV can be achieved by transient transfection of the plasmid set into 293T cells by the calcium phosphate method or from stable producer cell lines. Although proof of principle for the latter approach has been provided (Klages et al., 2000), it still suffers from limitations, e.g., for the production of SIN vectors. Unless very large amounts of a same vector are needed on a regular basis, transient production is still the method of choice for research purposes.
A. Cells
Cells (293T/17 from ATCC Cat. No. SD-3515) are probably the most critical factor for good titers. They need to be passaged every 2-3 days, as they start to form clumps that cannot be dissociated with one round of trypsin.
B. Solutions
- 0.5M CaCl2: Dissolve 36.75 g of CaCl2 and 2H2O (MW 147) (SigmaUltra Cat. No. C5080) into 500ml of H2O (distilled or double distilled). Store at -70°C in 50-ml aliquots. Once thawed, the CaCl2 solution can be kept at 4°C for several weeks without observing a significant change in the transfection efficiency.
- 2x HeBS: Dissolve 16.36g of NaCl (MW 58.44) (SigmaUltra Cat. No. S7653 (0.28M final), 11.9g of HEPES (MW 238.3) (SigmaUltra Cat. No. H7523) (0.05M final), and 0.213g of Na2HPO4, anhydrous (MW 142) (SigmaUltra Cat. No. S7907) (1.5mM final) into 800ml of H2O (distilled or double distilled). Adjust pH to 7.00 with 10M NaOH. Be careful, as obtaining a proper pH is very important. Below pH 6.95, the precipitate will not form, above pH 7.05, the precipitate will be coarse and transfection efficiency low. Then add H2O to 1000ml and make the final pH adjustment. Store at -70°C in 50-ml aliquots. Once thawed, the HeBS solution can be kept at 4°C for several weeks without observing a significant change in the transfection efficiency.
- Solution for mixing with plasmids: 50ml H2O distilled or double-distilled, 1M HEPES, pH 7.3 (Gibco- BRL, Cat. No. 15630-056) 125µl (2.5mM final). We have observed that the aspect and quality of the precipitates can vary among batches of distilled water. To circumvent this problem, we advise buffering distilled water that is used to dilute the plasmids. A final concentration of 2.5mM HEPES in the water will help maintain a proper pH and will not compete for the final pH with the HeBS, pH 7.00 (HEPES, pH 7.00, provided by HeBS is 25mM final, whereas HEPES, pH 7.3, provided by water is 0.625M final). Store at 4°C.
C. DNA
We use JetStar kits (Genomed GmbH, Germany) or Qiagen kits (Qiagen, GmbH, Germany) to prepare DNAs for transfection. In any case, the last step of the DNA prep should be an additional precipitation with ethanol (EtOH) and resuspension in 10mM Tris (SigmaUltra Cat. No. T6791) /1mM EDTA (SigmaUltra Cat. No. E6758) (TE 10/1). Do not treat DNA with phenol/chloroform as it may result in chemical alterations. Also, to avoid salt coprecipitation, we do not precipitate DNA below 20°C.
 |
| FIGURE 3 Phase-contrast photograph of 293T cells. 293T/17 cells are seeded the day before transfection at 1 to 3 million per 10-cm culture dish. At the time of transfection, cells must have the morphology and density as shown here. |
D. Transfection and Harvesting
The day before transfection, detach the 293T cells using trypsin/EDTA (Cat. No. 25300, GibcoBRL Life Technologies). Seed the 293T cells at 1 to 3 millions (cells must be approximately one-fourth to one-third confluent on the day of transfection) per 10-cm culture dishes (Cat. No. 353003, BD Biosciences) with 10ml of D10 complete medium composed of DMEM (Cat. No. 41966, GibcoBRL Life Technologies) supplemented with 10% fetal calf serum (FCS, Cat. No. 10099, Gibco- BRL Life Technologies), 1% penicillin-streptomycin (Cat. No. 15140, GibcoBRL Life Technologies), and 1% L-glutamine (Cat. No. 25030, GibcoBRL Life Technologies) (Fig. 3).
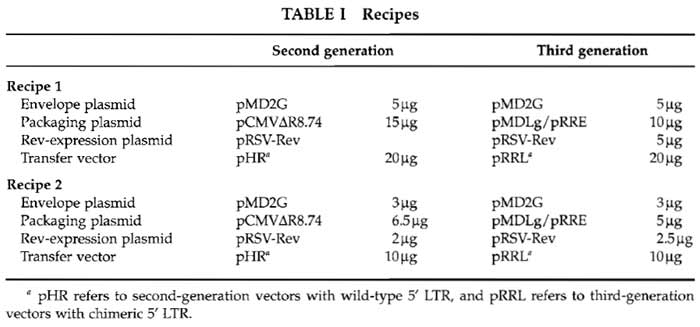
On day 0, in the evening, make the precipitate according to the recipes 1 (cf Table I for one 10-cm plate).
1 These recipes are the result of long optimizations in our laboratory and in the laboratory of Luigi Naldini and discussions with Antonia Follenzi.
 |
Adjust to 250µl with buffered water. Add 250µl of 0.5M CaCl2. Mix well and add this mix, dropwise, slowly (one drop every other second), on 500µl of HeBS 2×, while vortexing at maximum speed. Let stand still for 20 to 30min minimum (40 min max) on the bench. This time is critical for optimal formation of the precipitate. Excessive incubation times may induce the formation of a coarse precipitate, which is detrimental to transfection efficiency. A too short incubation will not induce the formation of a significant amount of precipitate, which will have no chance to develop further once diluted in the 10ml of culture medium. Also, it is advisory to incubate the solutions at room temperature or 37°C before mixing to standardize the precipitate characteristics. Add the precipitate slowly, dropwise on the cell monolayer. The plate should then be shaken gently but not stirred or all the precipitate will be in the center. At this point, the precipitate may not be visible, for it is too fine and will take several hours to sediment on the cells. If the precipitate is readily visible, it is too coarse and the transfection will most likely be less efficient.
On day 1 in the morning, check the cells. At this point, a very fine and sandy precipitate all over the plate should be seen, except on the cells and in their vicinity, possibly because cells absorb the CaPO4/DNA precipitate. Discard the medium after gentle, but firm, stirring (to eliminate the maximal amount of precipitate) and replace it with 10-15ml of fresh medium.
Notes: The medium must be prewarmed at 37°C because the 293T are very sensitive to thermal shock and can shrink and detach. Also, the medium must be added very gently to the cells. Even so, it is difficult not to make a hole in the monolayer.
On day 2, harvest the supernatant and replace with 10-15ml of medium as the day before. Spin the supernatant at 2500rpm for 10min at 4°C [in a tabletop centrifuge such as a Multifuge 3SR (Sorvall)], filter through a 0.45-µm PVDF filter (such as Millex- Durapore, Cat. No. SLHV 033, Millipore), and store at 4°C.
On day 3, harvest the supernatant, spin at 2500 rpm for 10min at 4°C, filter through a 0.45-µm filter, and pool with the supernatant of day 2. At this point, the pool of supernatants must contain approximately 106 transducing units (TU) per milliliter (as titered on HeLa cells, see later).
V. CONCENTRATION AND STORAGE
If a more concentrated lentivector suspension is required, the vector preparation needs to be concentrated. For concentration, we use Beckman Conical tubes (Cat. No. 358126, Beckman-Coulter) in a Surespin 630 rotor and a Discovery 90SE ultracentrifuge (Sorvall). Put 4-5 ml of 20% sucrose (SigmaUltra, Cat. No. S7903) in water at the bottom of the tube and fill up to the top with filtered supernatant, spin at 2600 rpm for 90 min and discard gently the supernatant by inversion. Let the tube dry inverted and resuspend the pellet (not always visible) with complete medium or serum-free medium, such as CellGro stem cell growth medium (Cat. No. 2001, CellGenix Technologie Transfer GmbH, Germany) if the subsequent experiments require the absence of serum.
The use of protein-containing medium is preferable over PBS to resuspend the viral pellet for two reasons: (1) the presence of proteins will stabilize the viral particles and (2) the surfactant effect of proteins will help redissolve the pellet.
VI. TITRATION
Titers of viruses in general and lentivectors in particular critically depend on the method and cells used for titration. The quantification of vector particles capable of achieving every step from cell binding to expression of the transgene depends on both vector and cell characteristics. First, the cell used as the target must be readily permissive to all steps from viral entry to integration of the vector genetic cargo. Second, the expression of the foreign gene must be monitored easily and rapidly reach levels sufficient for reliable quantification. Early vectors had the LacZ bacterial gene as the reporter under the control of the CMV promoter (Naldini et al., 1996). Current vectors now have the green fluorescent protein (GFP) gene as a reporter under the control of promoters that are active in most primary cells (Salmon et al., 2000). Measured titers can also vary with the conditions used for titration, i.e., volume of sample during vector-cell incubation, time of vector-cell incubation, and number of cells used. For several years now, we have been using HeLa cells as target. These cells are stable, easy to grow, and 100% susceptible to transduction by VSV-G-pseudotyped LVs. There are also now used commonly by many laboratories, which helps in comparing titers between laboratories.
A. Titration of Vectors in HeLa Cells by FACS
This method can only be used to titer stocks of vectors that carry a transgene that is easily monitored by FACS (such as GFP, any living colors, or any membrane protein that can be detected by flow cytometry) and whose expression is governed by a promoter that is active in HeLa cells (tissue-specific promotercontaining vector must be functionally assayed in specific cells), and titered by quantitative polymerase chain reaction (QPCR) in HeLa cells (see later).
We describe here the titration of a PGK-GFP vector. On day 0, seed Hela cells (Cat. No. CCL-2, ATCC) at 100k cells per well in a MW6 plate (Cat. No. 353224, BD Biosciences) in D10 complete medium. On day 1, put 500, 50, or 5 µl of the vector suspension (either pure from unconcentrated supernatants or diluted if it comes from a concentrated stock) in three independent wells. On day 2, remove the supernatant and replace with 2ml of fresh D10. On days 4 to 5, wash the cells with 2 ml of PBS without calcium-magnesium (Cat. No. 14190, Gibco-BRL Biosciences), detach them with 250µl of trypsin/EDTA (Cat. No. 25300, Gibco- BRL Life Technologies) for 1 min at 37°C, add 250 µl of 2% (w/v) formaldehyde (Cat. No. F8775, Sigma) in PBS without calcium-magnesium (to fix the cells, inactivate the trypsin and the vector particles), resuspend thoroughly, and analyze them for GFP expression using a flow cytometer (FACScan, Becton-Dickinson).
A reliable measure of the fraction of GFP+ cells relies on the level of GFP expression. In the example shown in Fig. 4, GFP-positive and GFP-negative cells can be readily discriminated when GFP is expressed from a human PGK promoter and allowed to accumulate in cells for 4 days. A marker can then be set to measure the fraction of transduced versus total cells.
In a typical titration experiment, only dilutions yielding 1-20% GFP positive should be considered for titer calculations. Below 1%, the FACS may not be accurate enough to reliably determine the number of GFP-positive cells. Above 20%, the chance for each GFP-positive target cell to be transduced twice increases significantly, resulting in an underestimation of the number of transducing particles. Once the appropriate dilution is chosen, apply the following equation:
titer (HeLa-transducing units/ml) = 100,000 (target HeLa cells) × (% of GFP-positive cells/100)/volume of supernatant (in milliliters).
![FIGURE 4 A representative FACS analysis of HeLa cells used for titration of GFP-coding LV. HeLa cells (105) were incubated with various volumes of a supernatant containing a LV expressing GFP under the control of the human PGK promoter [pRRLSIN.cPPT.PGK.GFP.WPRE (Follenzi <em>et al.</em>, 2000)] as described in the text. After 4 days, cells were detached, fixed, and analyzed by FACS for GFP fluorescence (x axis, four-decade log scale, FL1) versus number of cells (y axis, linear scale). The percentage of GFP-expressing cells was measured by placing a marker discriminating between GFP-negative (mean of fluorescence intensity 3-4) and GFPpositive (mean of fluorescence intensity 200) cells.](images/v1_pb_s14_c52_f04.jpg) |
| FIGURE 4 A representative FACS analysis of HeLa cells used for titration of GFP-coding LV. HeLa cells (105) were incubated with various volumes of a supernatant containing a LV expressing GFP under the control of the human PGK promoter [pRRLSIN.cPPT.PGK.GFP.WPRE (Follenzi et al., 2000)] as described in the text. After 4 days, cells were detached, fixed, and analyzed by FACS for GFP fluorescence (x axis, four-decade log scale, FL1) versus number of cells (y axis, linear scale). The percentage of GFP-expressing cells was measured by placing a marker discriminating between GFP-negative (mean of fluorescence intensity 3-4) and GFPpositive (mean of fluorescence intensity 200) cells. |
B. Total Vector Concentration Using Anti-p24 Immunoassay
Determination of total particle concentration is important to monitor the efficiency of vector production and packaging. One must keep in mind, however, that a high concentration of pelletable p24 viral capsid antigen can be measured from vector particles not containing any genomic RNA and/or devoid of envelope protein. Such an assay can thus not replace a transduction assay as described earlier and later. We currently use the p24 antigen capture assay provided by the AIDS vaccine program (Frederick, MD; email: schadent@mail.ncifcrf.gov). The minimal p24 concentration detected by this assay is 150pg/ml. HIV-1 or vector-containing supernatants are lysed by adding Triton X-100 (SigmaUltra Cat. No. T9284) to a final concentration of 1%. Lysed samples can be analyzed just after or stored at -20°C. Following the instructions provided by the manufacturer and using dilutions comprised between 1 / 10 and 1 / 10,000, we find that most of our vector preparations have a p24 concentration comprised between 100ng/ml and 1µg/ml. Given that most vector preparations have titers ranging from 5 × 105 to 107 Hela-TU/ml, this implies that the relative titer of our LV stocks is comprised between 1 and 20 Hela-TU/pg of p24. Of course, these results may vary depending on the packaging efficiency of a given transfer vector.
C. Titration of Vectors in HeLa Cells by Quantitative PCR
In the case of vectors coding for genes that cannot be detected readily by FACS or if the promoter is not active in HeLa cells, an alternative method is to measure the number of copies of LV stably integrated in HeLa target cells, after transduction as described earlier for GFP vectors. This assay, however, only measures the number of LV copies integrated in the target cell genome. The overall functionality of the vector must be tested at least once in cells in which the promoter is active and/or with appropriate techniques to detect the expression of the transgene product.
The QPCR assay proceeds as follows using a 7700 sequence detector (Applied Biosystems). HeLa cells are transduced as described earlier, but instead of being detached by trypsin, they are lysed with 200µl of lysis buffer in the plate and DNA is extracted using a DNAeasy kit (Qiagen GmbH, Germany). Then, 1- 2 µl of 200 µl total of DNA solution is analyzed for the copy number of HIV sequences using the following real-time PCR protocol.
1. Reaction
Always use filter tips. Mix for each sample and distribute in duplicate in a 96-well optical reaction plate (Cat. No. 4306737, Applied Biosystems): 12.5 µl 2× mix (Cat. No. RT-QP2X-03 Eurogentech, Belgium), 2.5 µl 10× oligonucleotide mix 1, 2.5 µl 10× oligonucleotide mix 2, 1-2 µl DNA sample, and up to 25 µl final with H2O. Close with optical caps (Cat. No. N801-0935, Applied Biosystems). Run a program appropriate for the probes, depending on the fluochrome used (FAM, VIC, TET, etc).
Notes: 10× oligonucleotide mixes are 1 µM probe and 3µM of each primer in water. Stocks of probes and primers usually come lyophilyzed and are stored at 10 µM in water. DNA ideally comes from 2 × 106 HeLa cells extracted and resuspended in 100µl (DNAeasy, Qiagen).
2. Oligonucleotides
Oligonucleotides can be ordered online from several companies such as Eurogentech or Sigma.
Oligonucleotides used to normalize for the amount of genomic DNA are specific for the β-actin (BAC) gene. The sequences are originally from a PE-Applied TAQMAN β-actin control reagent (Cat. No. 401846), which has been discontinued.
BAC-P (probe, sense) human β-actin
5'-(TET or VIC)- ATGCCCTCCCCCATGCCATCCTGCGT -(TAMRA)-3'
BAC-F (forward primer): TCACCCACACTGTGCCCATCTACGA
BAC-R (reverse primer): CAGCGGAACCGCTCATTGCCAATGG
Oligonucleotides for the amplification of HIV-1- derived vectors are specific for the 5' end of the gag gene (GAG). This sequence is present in all HIV-1 vectors for it is part of the extended packaging signal.
GAG-P (probe, antisense) HIV gag
5'-(FAM)-ACAGCCTTCTGATGTTTCTAACAGGCCAGG-(TAMRA)-3'
GAG-F (forward primer): GGAGCTAGAACGATTCGCAGTTA
GAG-R (reverse primer): GGTTGTAGCTGT CCCAGTATTTGTC
3. Analysis
An example of amplification profiles of HIV sequences in human DNA is given in Fig. 5 (as displayed by the ABI Prism program, Applied Biosystems). To analyze the amplification reaction, first set the threshold where the amplification curve is the steepest, both for the gene of interest (GAG-FAM, Fig. 5A) and for the internal control (BAC-VIC, Fig. 5B). Then, export the results as a Microsoft Excel sheet and draw a standard curve with standards of cells containing 1, 0.1, 0.01, and 0.001 copy of HIV per cell using the deltaCt values (Ct GAG minus Ct BAC). Ask Excel to display the formula (exponential). Apply the formula to unknown samples, it will give the HIV copy number of the corresponding sample. The titer of the supernatant can then be calculated as follows:
Titer (HeLa-transducing units/ml) = 100,000 (target HeLa cells) × number of copy per cell of the sample/volume of supernatant (in milliliters).
Notes: It is advisory to run a dual titration (FACS plus TAQMAN) using one GFP vector alongside the other vectors for each set of QPCR titration. This will help in comparing FACS titration with QPCR titration. A standard curve of HIV DNA in human DNA can also be made using a human cell line. In this case, keep in mind that the DNA content of these cell lines is different from the DNA content of normal primary human diploid cells. Small adjustments can be made using the table in Fig. 5D, which summarizes the chromosome counts of HeLa and CEM cells as provided in the ATCC catalog and gives calculated DNA contents for each cell.
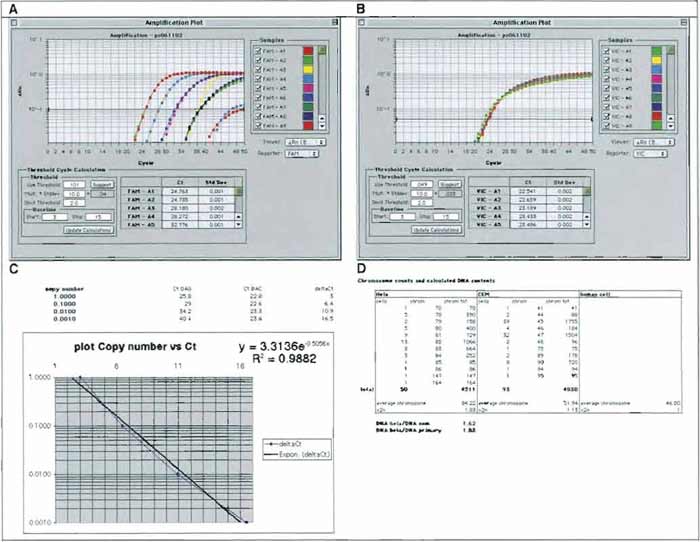 |
| FIGURE 5 A representative QPCR analysis used for titration of HIV-1-based LVs. DNA from 8E5 cells, a CEM-derivative (Cat. No. CRL-8993, ATCC; which contains a single copy of a replication defective HIV-1 provirus), was diluted by ten-fold increments in DNA from CEM cells. A sample of each dilution was submitted to QPCR amplification and monitoring using a Perkin-Elmer 7700 (Applied Byosystems) and sets of primers and probes specific for HIV gag sequences (GAG-FAM, A) or β-actin sequences (BAC-VIC, B). Amplification plots were displayed, and cycle threshold values (Ct) were set as described in the text. Values of GAG Ct and BAC Ct were exported in an Excel worksheet to calculate ΔCt values (x axis, linear scale) and plot them against copy number values (y axis, log scale) (C). The regression curve can then be used to calculate GAG copy numbers (Y value) of unknown samples by applying the formula to ΔCt values (X values) of the sample. (D) Calculated DNA contents of primary human cells, CEM, cells and HeLa cells based on the chromosome contents (after ATCC catalog). |
VII. TROUBLESHOOTING
In the case of lack of transduction of a specific cell type with a specific lentiviral vector, Fig. 6 helps in addressing most of the problems that could account for it.
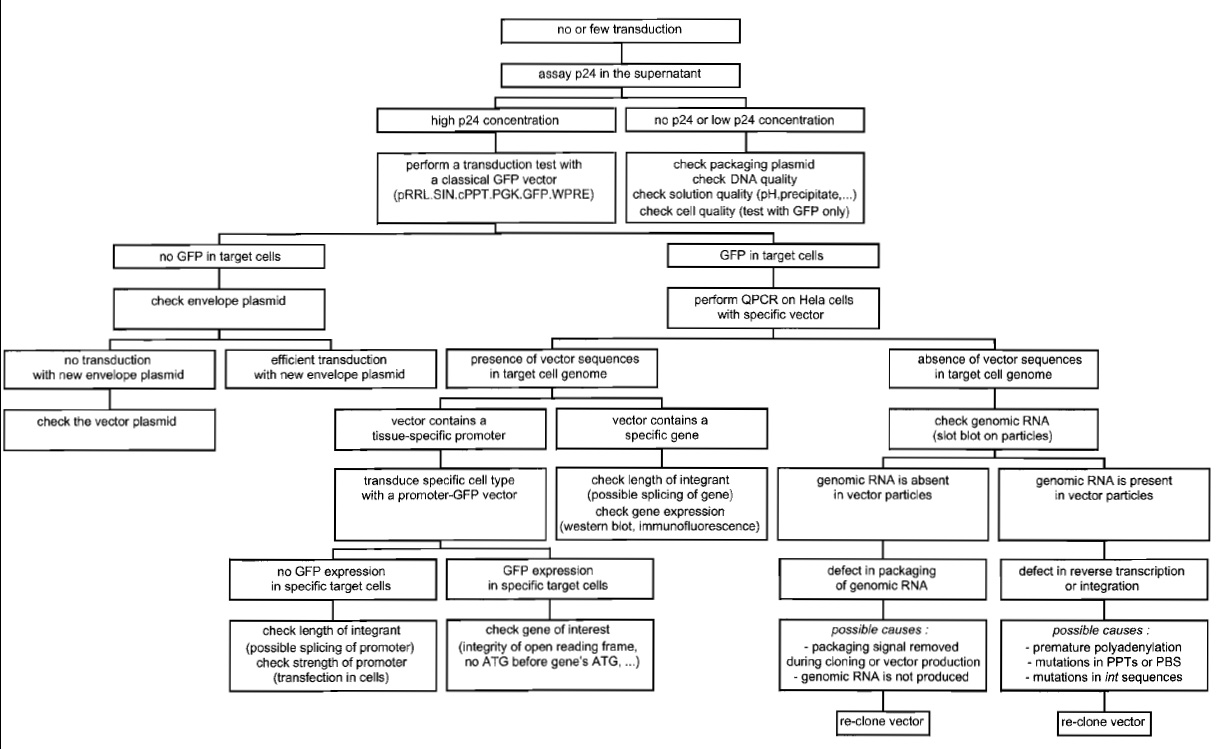 |
| FIGURE 6 Troubleshooting diagram for lentiviral vector production and transduction. |
VIII. CONCLUSION
Lentivector are vehicles of choice for the transduction of many primary cell targets, including neurons, retinal cells, pancreatic islet cells, lymphocytes, and hematopoietic stem cells. Although HIV-derived vectors have been best characterized, gene delivery systems have similarly been developed from other lentiviruses, such as simian (Negre et al., 2000), feline (Curran et al., 2000), and bovine (Berkowitz et al., 2001) immunodeficiency viruses and the equine infectious anemia virus (Mitrophanous et al., 1999). These other systems seem to have the same general properties as their HIV counterpart, but in human cells, HIVderived vectors appear to be more generally efficient, illustrating the fact that a virus adapts to its cognate target. From a biosafety standpoint, HIV-derived vectors may be safer than their nonhuman virus homologues. First, the genomic complexity of HIV is far greater than that of most other lentiviruses, including feline immunodeficiency virus and equine infectious anemia virus, which each have only six genes instead of the nine present in their human counterpart. Because in all cases a minimum of three genes, gag, pol, and rev, will likely be required for generating vector particles efficiently, the multiply attenuated HIV-based packaging system will be the farthest away from its parental virus. In addition, past experience with zoonoses teaches us that the pathogenicity of a given organism is largely unpredictable when it is transferred from its normal animal host into humans. Finally, millions of individuals worldwide have been screened for lentivirus-related diseases. No pathology has been associated with massively deleted forms of HIV-1; however, well-documented cases of longterm clinical nonprogression have occurred in patients infected with HIV-1 strains that carry genetic alterations far more subtle than those introduced in the third-generation HIV-1 packaging system.
Most recent developments of lentivector technology, such as its application for transgenesis (Lois et al., 2002) and RNA interference (Brummelkamp et al., 2002), indicate that the exploitation of this formidable tool is only beginning. Exciting times are ahead, in both experimental and therapeutic arenas.
Acknowledgments
We thank Antonia Follenzi for helpful discussions and Maciej Wiznerowicz for Fig. 2. Additional resources, such as vector sequences, are available at http://www.tronolab.unige.ch/. Downloading of QPCR oligonucleotide sequences and Excel QPCR calculation sheets can be made at http://www. medecine.unige.ch/~salmon/.
References
Berkowitz, R., Ilves, H., Lin, W. Y., Eckert, K., Coward, A., Tamaki, S., Veres, G., and Plavec, I. (2001). Construction and molecular analysis of gene transfer systems derived from bovine immunodeficiency virus. J Virol. 75, 3371-3382.
Brummelkamp, T. R., Bernards, R., and Agami, R. (2002). A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550-553.
Curran, M. A., Kaiser, S. M., Achaso, P. L., and Nolan, G. P. (2000). Efficient transduction of nondividing cells by optimized feline immunodeficiency virus vectors. Mol. Ther. 1, 31-38.
Dull, T., Zufferey, R., Kelly, M., Mandel, R. J., Nguyen, M., Trono, D., and Naldini, L. (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol. 72, 8463-8471.
Follenzi, A., Ailles, L. E., Bakovic, S., Geuna, M., and Naldini, L. (2000). Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nature Genet. 25, 217-222.
Klages, N., Zufferey, R., and Trono, D. (2000). A stable system for the high-titer production of multiply attenuated lentiviral vectors. Mol. Ther. 2, 170-176.
Lois, C., Hong, E. J., Pease, S., Brown, E. J., and Baltimore, D. (2002). Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868-872.
Mitrophanous, K., Yoon, S., Rohll, J., Patil, D., Wilkes, E, Kim, V., Kingsman, S., Kingsman, A., and Mazarakis, N. (1999). Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene Ther. 6, 1808-1818.
Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, E H., Verma, I. M., and Trono, D. (1996). in vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263-267.
Negre, D., Mangeot, P. E., Duisit, G., Blanchard, S., Vidalain, P. O., Leissner, P., Winter, A. J., Rabourdin-Combe, C., Mehtali, M., Moullier, P., Darlix, J. L., and Cosset, E L. (2000). Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 7, 1613-1623.
Salmon, P., Kindler, V., Ducrey, O., Chapuis, B., Zubler, R. H., and Trono, D. (2000). High-level transgene expression in human hematopoietic progenitors and differentiated blood lineages after transduction with improved lentiviral vectors. Blood 96, 3392-3398.
Sandrin, V., Boson, B., Salmon, P., Gay, W., Negre, D., Le Grand, R., Trono, D., and Cosset, E L. (2002). Lentiviral vectors pseudotyped with a modified RDl14 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 100, 823-832.
Zennou, V., Petit, C., Guetard, D., Nerhbass, U., Montagnier, L., and Charneau, P. (2000). HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 101,173-185.
Zufferey, R., Donello, J. E., Trono, D., and Hope, T. J. (1999). Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 73, 2886-2892.
Zufferey, R., Dull, T., Mandel, R. J., Bukovsky, A., Quiroz, D., Naldini, L., and Trono, D. (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 12, 9873-9880.
Zufferey, R., Nagy, D., Mandel, R. J., Naldini, L., and Trono, D. (1997). Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nature Biotechnol. 15, 871-875.
Support our developers




