In Situ Hybridization for Simultaneous Detection of DNA, RNA, and Protein
I. INTRODUCTIONFluorescence in situ hybridisation enables the detection of RNA and DNA in the cellular context, therefore allowing the study of gene expression at the single cell level. In situ hybridisation was described for the first time in 1969 in two independent studies (Gall and Pardue; 1969; John et al., 1969) that used radiolalelled probes and autoradiographic methods to detect abundant sequences. During the early eighties, nonisotopic probe-labelling methods were developed based on the introduction of haptens (such as biotin and digoxigenin) and fluorochrome-conjugated antibodies (for review, see Schwarzacher and Heslop-Harrison, 2000). The use of different probe labels and detection systems with multiple fluorochromes made possible the simultaneous detection of several target sequences in the same cell, including the detection of a specific gene and the corresponding pre-mRNA and/or mRNA (Custódio et al., 1999; Johnson et al., 2000; Xing et al., 1993,1995). A combination of fluorescence in situ hybridisation with immunofluorescence further allows the simultaneous visualisation of genes, transcripts, and proteins.
A major problem when conjugating protocols to simultaneously detect DNA, RNA, and proteins is to achieve good preservation of the different targets throughout the procedure and, at the same time, make them accessible for the probes. This article describes protocols that have been optimised over the years for the simultaneous detection of DNA, RNA transcripts, and proteins involved in RNA metabolism in the nucleus of mammalian cultured cell lines.
II. MATERIALS AND INSTRUMENTATION
35 × 10-mm (P35) tissue culture dishes (Sarstedt, Cat. No. 83.1800)
10 × 10-mm glass coverslips
Tweezers (style #7, sharp, hooked, Sigma, Cat. No. T 4912)
Water bath (variable temperature)
Heat block
Incubator at 37°C
Moist chamber (plastic box with lid containing some wet tissues inside)
Vacuum pump
Milli-Q ultrapure or bidistilled water
dNTPset, 100mM each dNTP ( Qbiogene, Cat. No. NTACG100)
Digoxigenin-11-dUTP, alkali stable (Roche, Cat. No. 1093088)
Dinitrophenyl(DNP)-11-dUTP (Molecular Probes, Cat. No. C-7610)
Alexa Fluor 488-5-dUTP (Molecular Probes, Cat. No. C-11397)
Fluorescein-12-dUTP (Roche, Cat. No. 137 3242)
BIOTAQ DNA polymerase (BIOLINE, Cat. No. BIO-21040)
Oligonucleotide probes with amino-modified bases (MWG Biotech)
Cy3 monoreactive dye (Amersham Biosciences, Cat. No. PA23001)
Cy5 monoreactive dye (Amersham Biosciences, Cat. No. PA2500)
Alexa Fluor 488 oligonucleotide amine-labelling kit (Molecular Probes, Cat. No. A-20191)
Escherichia coli tRNA (Sigma, Cat. No. R-4251)
Herring sperm DNA (Sigma, Cat. No. D-6898)
Human COT-1 DNA (Invitrogen, Cat. No. 152 79 011)
Mouse COT-1 DNA (Invitrogen, Cat. No. 184 40 016)
Dextran sulfate (Sigma, Cat. No. D-8906)
Albumin, bovine (Sigma, Cat. No. A-2153)
Formamide, molecular biology grade (Calbiochem, Cat. No. 344206)
AG 501-X8(D) resin (Bio-Rad, Cat. No. 142-6225)
Poly-L-lysine, 0.01% solution (Sigma, P4832)
Ribonucleoside-vanadyl complexes, 200mM (Sigma, Cat. No. R-3380)
Paraformaldehyde extra pure (Merck, Cat. No. 1.04005.1000)
Triton X-100 (Sigma, Cat. No. X-100)
Pepsin (Sigma, Cat. No. P-6887)
Gelatin from cold water fish skin (Sigma, Cat. No. G-7765)
Tween 20 (Sigma, Cat. No. P-9416)
Formamide p.a. (Merck, Cat. No. 1.09684)
Fluorescein-conjugated sheep antidigoxigenin (Roche, Cat. No. 1 333 062)
Alexa Fluor 488-conjugated goat antifluorescein (Molecular Probes, Cat. No. A-11090)
Cy3-conjugated mouse antidigoxin (Jackson Immuno-Reseach Labs, Inc., Cat. No. 200-162-156)
Cy5-conjugated mouse antidigoxin (Jackson Immuno-Reseach Labs, Inc., Cat. No. 200-172-156)
FITC-conjugated rabbit anti-DNP (Molecular Probes, Cat. No. A-6423)
Rabbit anti-DNP (Molecular Probes, Cat. No. A-6430)
Alexa Fluor 488-conjugated goat antirabbit (Molecular Probes, Cat. No. A-11034)
TRITC-conjugated donkey antirabbit (Jackson Immuno- Reseach Labs, Inc., Cat. No. 711-025-152)
Other fluorochrome-conjugated secondary antibodies (Jackson ImmunoReseach Labs, Inc.)
Formaldehyde solution min. 37%, p.a. (Merck, Cat. No. 1.04003.1000)
Vectashield mounting medium (Vector, Cat. No. H- 1000)
Microscopy glass slides
Nail polish
III. PROCEDURES
A. Labeling of Probes
There are several methods to label DNA probes. Choosing a specific method depends on the kind of probe to be labelled. For example, whole recombinant plasmids or purified inserts are easily labelled by nick translation, whereas it is more advantageous to label shorter DNA fragments (100-600bp) by polymerase chain reaction (PCR). Oligonucleotides can be synthesised with amino-modified nucleotides (amino-allyl thymine) at specific positions in the sequence and labelled by conjugation with activated fluorophores.
1. Labelling of Probes by PCR
Solutions
- Sodium acetate 3M, pH 5.5. Weigh 40.81g of NaCH3COOH to 100ml of solution. Adjust the pH to 5.5 with acetic acid. Sterilise by autoclaving.
- 10mg/ml E.coli tRNA. Dissolve 0.1g of tRNA in 10ml of sterile water. Aliquot and store at -20°C.
Steps
- Make up the following PCR reaction:
Sterile water to bring the final volume (including enzyme) to 50µl
5µl 10 × PCR buffer (supplied with the enzyme)
1.5µl 50mM MgCl2 (the optimal concentration of MgCl2 must be determined empirically; initially try 1.5mM, taking into account that some PCR buffers already contain MgCl2)
1µl 1 ng/µl template DNA
2µl primer mix, 10µM (10pmol/µl) each
1µl 10mM dATP
1µl 10mM dCTP
1µl 10mM dGTP
3.75µl 2mM dTTP
2.5µl 1mM dig-11-dUTP (or other modified nucleotide)
2.5 units Taq DNA polymerase
The ratio dTTP :modified nucleotide in the previous mixture is 3:1. This ratio is ideal for generating highly labelled probes necessary to detect target sequences present at a low copy number. To generate moderately labelled probes the ratio of dTTP :modified nucleotide in the PCR reaction should be increased. - Mix the reagents and centrifuge briefly to collect the mixture into the bottom of the tube.
- Place the reaction in a termocycler and perform
the PCR under the following conditions:
Denaturation before the first cycle: 3 min at 95°C.
30 cycles of denaturation for 30s at 95°C annealing for 30s at 60°C and elongation for lmin at 72°C.
Final elongation: 10min at 72°C.
Cycling conditions will depend on the template and primers used. Other cycling conditions may be required to get optimal results. - Stop the reaction by chilling at 4°C. Run 5µl of the reaction on an agarose gel and estimate the amount of product generated in the PCR reaction.
- Add water to adjust the volume of the PCR reaction to 100µl and purify the labelled probe by ethanol precipitation: add 5µl of sodium acetate 3M, pH 5.5, 1 µl of 10mg/ml E.coli tRNA (this will act as a carrier RNA to help precipitation), and 200µl of cold 100% ethanol (-20°C).
- Incubate for 15min at -70°C or for 1h at -20°C to allow precipitation of the labelled fragments.
- Centrifuge at 13,000rpm for 15min at 4°C. Remove the supernatant and add 200µl of cold 70%(v/v) ethanol. Centrifuge for 5min in the same conditions.
- Discard the supernatant completely and air dry the pellet. Dissolve the pellet in sterile water to achieve a concentration of probe of 10ng/µl. Store at -20°C.
2. Labelling of Modified Oligonucleotide Probes by Conjugation with Activated Fluorophores
The oligonucleotide probes should be at least 40-50 nucleotides long to minimize the possibility of unspecific binding to nontarget cellular RNAs. When choosing the sequences, avoid regions that may form highly stable secondary structures or dimmerise. If more than one oligonucleotide is used in the same hybridisation, they all should have similar GC content. The oligonucleotides should be synthesised with four or five thymines in the sequence changed to amino-allyl thymine. The modified nucleotides should be at least 10 bases apart to prevent quenching (Long et al., 1995; Singer, 1998). The amino-modified thymine can then be coupled chemically to fluorophores as described.
Solutions
- Carbonate buffer: 0.1M NaHCO3, pH 8.8. Weigh 0.84 g of NaHCO3 to 100ml of solution. Adjust the pH with NaOH. Make aliquots and store at -20°C.
- Fluorophore solution: Dissolve one vial of the activated fluorophore (Cy3 monoreactive dye, Cy5 monoreactive dye, or Alexa Fluor 488 from the oligonucleotide amine-labelling kit) in 30µl of dimethyl sulfoxide. Store at -20°C.
- TE, pH 8.0 (10mM Tris, 1mM EDTA, pH 8.0): Add 1ml of 1M Tris, pH 8.0, and 0.2ml of 0.5M EDTA. Adjust the volume to 100ml with water. Sterilise by autoclaving.
Steps
- In a microtube, dilute 5µg of the modified oligonucleotide (higher oligonucleotide concentration will decrease the yield of the labelling reaction) with carbonate buffer to a final volume of 35µl (buffers containing primary amino groups, such as TRIS, should be avoided as they inhibit the conjugation reaction).
- Add 15µl of fluorophore solution and mix well by vortexing. Allow the labelling reaction to proceed for 24h in the dark at room temperature.
- Add sterile water to adjust the volume of the labelling reaction to 100µl and purify the labelled oligonucleotide by two rounds of ethanol precipitation as follows. Add 10µl of 3M sodium acetate, pH 5.5, 1µl of 10mg/ml E.coli tRNA, and 300µl of cold 100% ethanol (-20°C). Incubate overnight at -20°C to allow precipitation of the labelled oligonucleotide probe.
- Centrifuge at 13,000rpm for 15min at 4°C. Remove the supernatant and wash with 200µl of cold 100% ethanol (do not use 70% ethanol as it will dissolve the precipitated oligonucleotide). Centrifuge for 5 min in the same conditions.
- Remove the supernatant and dissolve the pellet in 100µl of sterile water.
- Reprecipitate by adding 10µl of 3M sodium acetate, pH 5.5, and 300µl of cold 100% ethanol and incubate for 4h at -20°C.
- Repeat step 4, then discard the supernatant completely, and air dry the pellet. Dissolve the labelled oligonucleotide in 100µl of sterile water to achieve a final concentration of 50ng/µl (assuming no losses during the procedure).
B. Preparation of Hybridization Mixture
Solutions
- 20 × SSC (saline sodium citrate): 0.3M sodium citrate, 3M NaCl, pH 7.4. Weigh 88.24g of sodium citrate and 175.32 g of NaCl for 1 liter of solution. Sterilise by autoclaving.
- 50% dextran sulfate: Dissolve 5 g of dextran sulfate in 4ml of water. Adjust the volume to 10ml and sterilise by autoclaving. Store at 4°C. Heat at 55°C and mix well before taking an aliquot.
- 0.5M sodium phosphate buffer, pH 7.0: 0.31M NaH2PO4 and 0.19M Na2HPO4. Weigh 1.84 g NaH2PO4 and 1.37g Na2HPO4 for 50ml of solution. Adjust the pH to 7.0 with 10M NaOH if necessary. Sterilise by autoclaving.
- 10% bovine serum albumin (BSA): Dissolve 1g of BSA in 7ml of sterile water. Adjust the volume to 10 ml. Aliquot and store at -20°C.
- 1mg/ml sonicated herring sperm DNA (100-200 bp). Dissolve 0.1 g of herring sperm DNA in 10ml of sterile water. Sonicate until the average fragment size is 100-200bp (control the fragment size on agarose gel electrophoresis). Measure ODs at 260nm to determine the concentration of the DNA and dilute to 1mg/ml. Aliquot and store at -20°C.
- Deionized formamide: Add 5g of ion-exchange resine to 100ml of molecular biology grade formamide. Stir for 3h, wait for the resine to settle down, and filter through 0.22-µm or Whatmann No. 1. Aliquot and store at -20°C.
- 50% formamide hybridization buffer: 50% formamide, 2 x SSC, 10% dextran sulfate, and 50mM sodium phosphate, pH 7.0. Add 5 ml of deionised formamide, 2ml of 50% dextran sulfate, 1ml of 20 x SSC, and 1ml of 0.5 M sodium phosphate buffer, adjust the pH to 7.5 with 1N HCl, and add sterile water to 10ml. Mix well, aliquot, and store at -20°C.
- 20% formamide hybridization buffer: 20% formamide, 2 x SSC, 10% dextran sulfate, 0.2% BSA, and 1µg/µl tRNA, pH 7.0. For 1ml, add 200µl of deionised formamide, 200µl of 50% dextran sulfate, 100 µl of 20 x SSC, 20µl of 10% BSA, 100 µl of 10 mg/ml tRNA, and 380µl of sterile water. Mix well and store at 4°C.
1. PCR or Nick Translation-Labeled Probes
Long genomic cloned sequences can be used as probes, but they often contain repetitive sequences that can produce a high background due to crosshybridization. To reduce this background, a suppressive hybridisation must be performed. In this case, an excess of unlabelled DNA enriched in repetitive sequences (e.g., COT-1 DNA and herring sperm DNA) is included in the hybridization mixture. For probes that do not contain repetitive sequences, a nonsuppressive hybridization is performed. In this case, tRNA is used as a carrier for probe precipitation. Steps
- For suppressive hybridization, add per hybridisation 10-100ng of labelled probe, 5.6µg of COT-1 DNA, and 2.4 µg of sonicated herring sperm DNA. For nonsuppressive hybridization, add 8 µg of E.coli tRNA to the labelled probe (the amount of probe in the hybridization must be determined empirically).
- Precipitate by adding 0.05 volumes of 3M sodium acetate, pH 5.0, and 2 volumes of cold ethanol (-20°C). Incubate for 15 min at -70°C or 1h at -20°C
- Centrifuge at 13,000rpm for 15min at 4°C. Remove the supernatant and wash with cold 70%(v/v) ethanol. Centrifuge for 5 min in the same conditions.
- Discard the supernatant completely and air dry the pellet. Dissolve the pellet in 50% formamide hybridization buffer (use 8µl per hybridization) and store at -20°C. Note that before taking an aliquot of the hybridisation buffer this must be warmed up to 37°C and vortexed to homogenise all the components.
2. Oligonucleotide Probes
Prepare the hybridization mixture by diluting the labelled oligonucleotide in the 20% formamide hybridization buffer to the appropriate concentration (1-2ng/µl of each oligonucleotide). If the oligonucleotide stock solution is too diluted, dry it in the Speed-Vac and then dissolve in the hybridization buffer. C. Preparation of Cells
Solutions
- 10× phosphate-buffered saline (PBS): 1.37M NaCl, 26.8mM KCl, 80.6mM Na2HPO4, 14.7mM KH2PO4. Weigh 80g NaCl, 2g KCl, 11.44g Na2HPO4, and 2g KH2PO4 for 1 liter of solution. Sterilise by autoclaving.
- 7.4% paraformaldehyde (2 × PFA): Weigh 7.4g of PFA into a glass flask, add water to 100ml, and stir at 60°C for severa1hours with the flask closed. Add 100µl of 1M NaOH dropwise and keep stirring until the solution is completely clear (pH should be around 7.5). Let it cool down to room temperature and filter through a 0.22-µm filter. Store at -20°C in 5-ml aliquots. Thaw at 60°C and use within 1-2 days.
- 3.7% formaldehyde in PBS: Add 5ml of 2 x PFA, 1ml of 10 x PBS, and sterile water to 10ml.
- 3.7% formaldehyde/5% acetic acid/0.9% NaCl: Add 5 ml of 2 x PFA, 0.5 ml of acetic acid, 0.3 ml 5 M NaCl, and sterile water to 10ml.
- 10% (w/v) Triton X-100: Weigh 1 g of Triton X-100 and add sterile water to 10ml. Dissolve by warming to 37°C. and vortexing.
- 0.5% Triton X-100 in PBS with 2mM vanadyl-ribonucleoside complex: Add 0.5ml of 10% Triton X-100, 1ml 10 x PBS, 100µl of 200mM vanadyl-ribonucleoside complex, and sterile water to 10ml.
- 0.01% (w/v) pepsin in 0.01M HCl. Prepare immediately before use with HCl at 37°C. Dissolve 5mg of pepsin in 50µl of 0.01M HCl and then adjust the volume with 0.01M HCl to 50ml.
Steps
All the procedures are performed with cells attached on 10 × 10-mm glass coverslips placed in 35 x 10-mm tissue culture dishes. One dish can accommodate four of these coverslips, and 1ml of solution is enough for each fixation, permeabilisation, and washing step.
- Grow monolayer cells to subconfluent density directly on 10 × 10-mm coverslips and wash them in serum-free medium. Alternatively, wash suspension cells in serum-free medium and allow them to adhere onto 10 × 10-mm poly-L-lysine-coated glass coverslips for 1 min at room temperature (approximately 2 × 105 cells in 30µl of medium per coverslip).
- Fix and permeabilize the cells according to one
of the following protocols.
- Fix with 3.7% formaldehyde in PBS for 10min at room temperature, wash 3 × 5 min with PBS, permeabilise with 0.5% Triton X-100 in PBS with 2mM vanadyl-ribonucleoside complex for 10 min, and wash again 3 × 5 min with PBS. Proceed to protocols D, E, or F (when conjugating more than one protocol, go sequentially from D to E to F).
- Fix with 4% formaldehyde, 5% acetic acid, and 0.9% NaCl for 20min at room temperature, wash 4 × 5min with PBS (include 2mM vanadyl-ribonucleoside complex in the last wash), and leave in 70% ethanol at -20°C for at least 1h. Before hybridization, rehydrate the cells in PBS and digest with 0.01% pepsin in 0.01 M HCl for 5 min at 37°C. Rinse with PBS and inactivate the protease by fixation with 3.7% formaldehyde in PBS for 5 min at room temperature. Wash 3 × 5 min with PBS and proceed to protocol D or E (cannot be used with protocol F).
Step 2b allows a better access of the probe to the target RNA sequences in the cell. Therefore, this protocol may be required when hybridizing with oligonucleotide probes that detect the splice junctions of mRNA. Note that monolayer cells may detach from the coverslip during treatment with pepsin.
D. Hybridization to RNA
Solutions
- 20% (w/v) Tween 20: Weigh 2 g of Tween 20 and add sterile water to 10ml. Dissolve by warming to 37°C and vortexing.
- 20% (w/v) gelatin from cold water skin fish: Weigh 0.2 g of gelatin and add sterile water to 1ml. Mix well, aliquot, and store at -20°C.
- 50% formamide, 2 × SSC, 0.01% Tween 20, pH 7.0: Add 5 ml of formamide, 1ml of 20 x SSC, 5 µl of 20% Tween 20, and sterilile water to 10ml. Adjust the pH to 7.0 with 1M HCl (use deionised formamide and add 2 mM vanadyl-ribonucleoside complex for prehybridisation incubation and formamide p.a. in posthybridisation washes).
- 20% formamide, 2 × SSC, 0.01% Tween 20, pH 7.0: Add 2ml of formamide, 1ml of 20 x SSC, 5 µl of 20% Tween 20, and sterile water to 10ml (use deionised formamide and add 2mM vanadyl-ribonucleoside complex for prehybridization incubation and formamide p.a. in posthybridization washes).
- 2 × SSC, 0.01% Tween 20: Add 1ml of 20 x SSC, 5µl of 20% Tween 20, and water to 10ml.
- 4 × SSC, 0.1% Tween 20: Add 100ml 20 x SSC, 2.5 ml 20% Tween 20, and sterile water to 500ml.
- 4 × SSC, 0.1% Tween 20, 1% BSA, 0.2% gelatin: To 1ml add 200 µl20 x SSC, 5µl20% Tween 20, 100 µl 10% BSA, 10µl 20% gelatin, and 685 µl of sterile water.
Steps
- Denature double-stranded DNA probes by heating the hybridization mixture at 75°C for 5min (use 8 µl per coverslip). Place on ice for nonsupressive hybridization or at 37°C for 10min for suppressive hybridization to allow preannealing of repetitive DNA sequences.
- Incubate the cells in 50% formamide, 2 x SSC, 0.01% Tween 20, and 2mM vanadyl-ribonucleoside complex for 5min at 37°C (when hybridizing with oligonucleotides, use 20% formamide).
- Aspirate the solution as much as possible, especially around the coverslip, without allowing the cells to dry. Place 8 µl of hybridisation mixture in the center of the coverslip and incubate at 37°C in a moist chamber for 3-16h (1-3h for oligonucleotides).
- Perform posthybridisation washes at the desired
stringency. The washes described here are just a guideline.
For optimal results, the percentage of formamide,
salt concentration, and temperature should be optimized
for each probe. Wash sequentially as follows:
Wash I: 3 × 5 min at 45°C with 50% formamide p.a., 2 x SSC, and 0.01% Tween 20
Wash II: 3 × 5 min at 45°C with 2 x SSC and 0.01% Tween 20
When hybridizing with oligonucleotides, the washes should be less stringent, e.g., wash at 37°C or at 42°C with only 20% formamide in wash I. Perform washes with all the solutions prewarmed at the desired temperature. This step can be performed either in an incubator or in a water bath with the dishes inside a floating metal platform. - Proceed to step 7 if the probe is labelled directly with a fluorescent dye and no amplification step is required. Alternatively, wash with 4 x SSC and 0.1% Tween 20 for 5 min at 37°C and proceed to step 6 for detection with the appropriate antibodies.
- Aspirate the solution, place 5µl of diluted antibody in the center of the coverslip, and incubate for 30min at 37°C in a moist chamber. Following each incubation step wash 3 × 5 min with 4 x SSC and 0.1% Tween 20 at 37°C. For digoxigenin-labelled probes, use fluorescein-conjugated sheep antidigoxigenin (1/100) followed by Alexa Fluor 488-conjugated goat antifluorescein (1/200) if an amplification step is required. Alternatively, use Cy3- or Cy5-conjugated mouse antidigoxin (1/250). For DNP-labelled probes, use FITC-conjugated rabbit anti-DNP or rabbit anti-DNP followed by Alexa Fluor 488-conjugated goat antirabbit or TRITC-conjugated donkey antirabbit. Dilute antibodies in 4 x SSC, 0.1% Tween 20, 1% BSA, and 0.2% gelatin.
- Wash briefly in PBS, fix the signal with 3.7% PFA in PBS for 10min at room temperature, and wash 3 x 5 min in P BS.
- Proceed to protocols E or F or mount in a microscopy glass slide with Vectashield mounting medium and seal with nail polish.
E. Hybridization to DNA
Solutions
- 70% formamide, 2 × SSC, 50mM sodium phosphate, pH 7.0: Add 7 ml of deionized formamide, 1ml of 20 x SSC, 1ml of 0.5 M sodium phosphate buffer, and sterile water to 10ml. Adjust the pH to 7.0 with 1M HCl.
- 50% formamide, 2 × SSC, 50mM sodium phosphate, pH 7.0: Add 5 ml of deionized formamide, 1ml of 20 x SSC, 1ml of 0.5 M sodium phosphate buffer, and sterile water to 10ml. Adjust the pH to 7.0 with 1M HCl.
Steps
- Denature the double-stranded DNA probes by heating the hybridization mixture at 75°C for 5min (use 8 µl per coverslip). Place on ice for nonsupressive hybridization or at 37°C for 10min for suppressive hybridization to allow preannealing of the repetitive DNA sequences.
- Incubate the cells in 2 x SSC at 40°C for 5min (this step is to prewarm the cells and avoid lowering the temperature in the denaturation step).
- For denaturation, incubate at 73°C in 70% formamide, 2 x SSC, and 50mM sodium phosphate, pH 7.0, for 3 min followed by 50% formamide, 2 x SSC, and 50mM sodium phosphate, pH 7.0, for 1 min.
- Aspirate the solution as fast as possible and place 8 µl of hybridization mixture in the center of the coverslip. Incubate for 16 h at 37°C in a moist chamber.
- Perform posthybridization washes at the desired
stringency. The washes described here are just a guideline.
For optimal results, the percentage of formamide,
salt concentration, and temperature should be optimized
for each probe. Wash sequentially as follows:
Wash I: 3 × 5 min at 45°C with 50% formamide p.a., 2 x SSC, and 0.01% Tween 20
Wash II: 3 × 5 min at 45°C with 0.5 x SSC and 0.01% Tween 20 - Proceed to step 8 if the probe is directly labelled with a fluorescent dye and no amplification step with antibodies is required. Alternatively, wash with 4 x SSC, 0.1% Tween 20 for 5min at 37°C and proceed to step 7 for detection with the appropriate antibodies.
- For detection of hybridization sites, follow step 6 of protocol D. When conjugating several protocols it is essential to avoid cross-reactivity between the antibodies used in the different protocols and to choose different fluorescent dyes for each detection.
- Wash briefly in PBS, fix the signal with 3.7% PFA in PBS for 10min at room temperature, and wash 3 x 5 min in PBS.
- Proceed to protocols F or mount in a microscopy glass slide with Vectashield mounting medium and seal with nail polish.
F. Detection of Proteins by Indirect Immunofluorescence
Solutions
- PBS, 0.05% Tween 20: To 500ml add 50ml 10 x PBS, 1.25 ml 20% Tween 20, and 450ml of sterile water.
- PBS, 0.05% Tween 20, 0.2% gelatin: Add 10µl of 20% gelatin to 1ml of PBS and 0.05% Tween 20.
- 2% formaldehyde in PBS: Prepare by adding 2.7ml of formaldehyde solution at 37% to 50ml of PBS.
Steps
- Wash cells with PBS, 0.05% Tween 20 for 5 min.
- Incubate with the primary antibody (diluted in PBS, 0.05% Tween 20, 0.2% gelatin) for 1h at room temperature in a moist chamber.
- Wash 3 × 5 min in PBS, 0.05% Tween 20 at room temperature.
- Incubate with the appropriate secondary antibody (diluted in PBS, 0.05% Tween 20, 0.2% gelatin) for 30min at room temperature in a moist chamber.
- Wash 3 × 5min in PBS, 0.05% Tween 20 at room temperature.
- Wash briefly in PBS. Fix with 2% formaldehyde in PBS for 10min.
- Wash 3 × 5 min in PBS, mount in a microscopy glass slide with Vectashield mounting medium, and seal with nail polish. For visualization of β-globin gene expression, see Fig. 1.
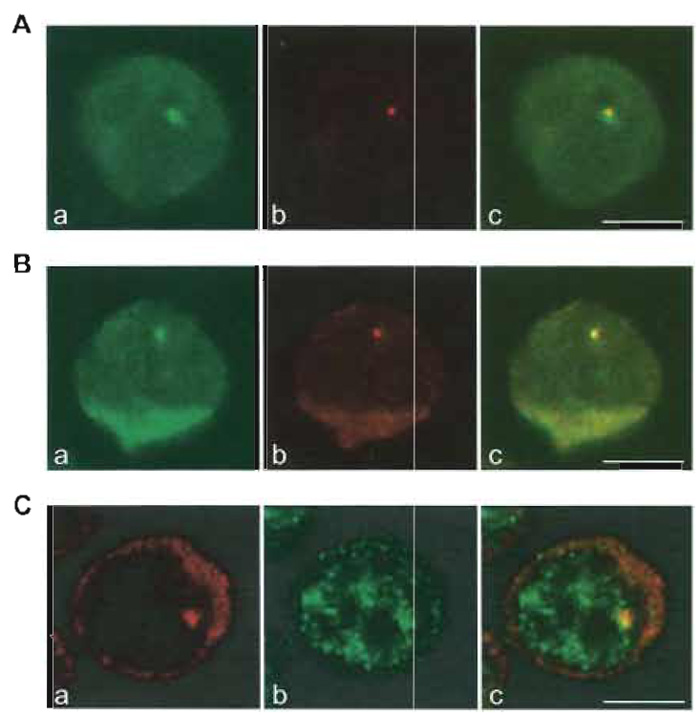 |
| FIGURE 1 Visualization of β-globin gene expression at the single cell level. (A) Simultaneous detection
of human β-globin RNA and DNA. Murine erythroleukaemia (MEL) cells were stably transfected with
multiple copies of the human β-globin gene cloned in the micro-LCR expression vector (Collis et al., 1990;
Cust6dio et al., 1999). After induction of β-globin gene expression, cells were fixed with formaldehyde/acetic
acid, treated with pepsin, and hybridised with a probe complementary to the transcribed sequence of the
gene, labeled with digoxigenin (a, signal detected with fluorescein). A bright fluorescent focus is detected in
the nucleus. Following fixation of the RNA signal with formaldehyde, cells were denatured and hybridized
with a probe that recognizes the micro-LCR, labelled with DNP (b, signal detected with Texas red). A fluorescent
focus in the nucleus indicates the integration site of the transfected gene locus. Overlay of the green
and red images in c shows colocalization of the two focal signals, indicating that the human β-globin RNA
is detected in the nucleus at the site of transcription. (B) Simultaneous detection of human β-globin nascent
transcripts and spliced mRNA. MEL cells expressing the human β-globin gene were fixed with formaldehyde/acetic acid, treated with pepsin, and hybridized sequentially to detect human β-globin nascent transcripts
and spliced mRNA. The cells were first hybridized with a mixture of two splice junction
oligonucleotide probes (SJ I/II probe complementary to the last 12 nucleotides of exon I and the first 12
nucleotides of exon II; SJ II/III complementary to the last and first 12 nucleotides of exons II and III, respectively)
labeled with DNP (a, signal detected with fluorescein). Cells expressing the human β-globin gene for
4 days contain a fluorescent focus in the nucleus and additional cytoplasmic staining. Following fixation of
the signal with formaldehyde, cells were incubated with a probe that hybridizes to the full-length human β-globin RNA, labeled with digoxigenin (b, signal detected with rhodamine). (c) A superimposition of the two
images. The focal signals colocalize in the nucleus, indicating that splicing of β-globin pre-mRNA is taking
place in close proximity to the site of transcription. (C) Simultaneous detection of human β-globin transcripts and an RNA processing factor. MEL cells expressing the human β-globin gene were fixed with formaldehyde, permeabilised with Triton, and hybridized with a probe that spans the transcribed sequence of the human β-globin gene (a, signal detected with Cy3). RNA is detected in the nucleus as a focus and in the cytoplasm. After fixation of the RNA signal with formaldehyde, cells were incubated with an antibody against the splicing coactivator SRm160 (rAb-SRm160; Blencowe et al., 1998) (b, signal detected with Alexa Fluor 488). SRm160 labels the nucleoplasm with a speckled pattern. Overlay of the two signals (c) shows colocalization of the nuclear RNA signal with a speckle of SRm160, indicating recruitment of this protein to the site of human β- globin RNA transcription and splicing. Bars: 5µm. |
IV. COMMENTS
The methods described in this article can be used to detect RNA and DNA, RNA and protein, DNA and protein, or RNA, DNA, and protein. In double RNA/DNA hybridization, hybridization to RNA is always performed first (without DNA denaturation, RNA is the only target for the probe). Then, the cellular DNA is denatured and hybridization to DNA is performed. The ideal probe for DNA detection should target nontranscribed regions of the genome and should not produce a signal if hybridised without prior denaturation of cellular DNA. The same probe, labelled with different reporters, can alternatively be used to simultaneously detect a DNA region and the corresponding transcripts. In this case, an RNase treatment must be introduced after the detection of the probe hybridized to RNA (Xing et al., 1995). An alkaline denaturation of DNA can also be performed instead of heat denaturation because it simultaneously degrades the cellular RNA (see Johnson et al., 2000). To minimise loss of the RNA signal during DNA denaturation, the probe hybridized to RNA should be detected with two layers of antibodies and fixed with formaldehyde. Direct labelling of the probe with fluorescent dyes without any step of signal amplification with antibodies is not recommended for this procedure. For multiple hybridisation and immunofluorescence experiments, different labels for the probes and fluorescent dyes coupled to antibodies must be combined. Particular attention should be taken to avoid cross-reactivity between antibodies used at different steps. For example, if a mouse antibody is used in detection of the RNA signal, antimouse immunoglobulin antibodies cannot be used either in the detection of the DNA signal or in immunofluorescence. When combining in situ hybridization with protein detection, the immunofluorescence is usually performed after the hybridization steps. This is especially important when conjugating RNA and protein detection because primary antibodies used for immunofluorescence are often contaminated with RNases, resulting in a significant decrease of the RNA signal. However, some epitopes are no longer recognised by the antibodies after incubation of the cells with formamide, especially after a denaturation step. In this case, immunofluorescence must be performed first and the signal fixed before hybridization. An important control to guarantee that the pattern of the antibody labelling is not changed by formamide is to stain cells using standard immunofluorescence conditions and to compare the pattern with the one obtained after RNA or DNA hybridization. The methods presented here were partly adapted from an original protocol reported by Zirbel et al., (1993). The pepsin treatment was introduced for the specific detection of splice junctions on nascent RNA transcripts in the nucleus (Wijgerde et al., 1995).
V. PITFALLS
Appropriate negative controls are essential to distinguish specific signals from fluorescent noise due to unspecific hybridization. For RNA hybridization the best control is to use cells that do not transcribe the target RNA. To overcome unspecific hybridisation, the stringency of the hybridization and posthybridization washes can be increased until no signal is obtained in the negative control cells, but without eliminating the signal in the cells that express the target RNA. If this is not possible, a new probe should be designed and tested in the same way. Control probes are also very important, especially when very small oligonucleotide probes are used. One example is splice junction probes. These probes are designed to hybridise with the last nucleotides of one exon and the first nucleotides of the following exon and to produce a stable hybrid only if the two halves of the probe hybridize. An appropriate control probe should be designed to exclude that only half of the probe is capable of producing a stable hybrid. Other negative controls for RNA hybridization include mock hybridization (no probe in the hybridization mixture) and RNase treatment before hybridization (0.1 mg/ml DNase-free RNase in 2 x SSC at 37°C for 1h). Additional controls should be performed when using probes that contain plasmid sequences. Namely, the plasmid backbone (without insert) should be labelled and used as a probe. If this produces a signal (which happens frequently if the cells have been transfected with related plasmids), the insert should be isolated from the recombinant plasmid before labelling.
References
Blencowe, B. J., Issner, R., Nickerson, J. A., and Sharp, E A. (1998). A coactivator of pre-mRNA splicing. Genes Dev. 12, 996-1009.
Collis, E, Aneoniou, M., and Grosveld, E (1990). Definition of the minimal requirements within the β-globin gene and the dominant control region for high level expression. EMBO J. 9, 233-240.
Custódio, N., Carmo-Fonseca, M., Geraghtly, E, Pereira, S. H., Grosveld, E, and Antoniou, M. (1999). Inefficient processing impairs release of RNA from the site of transcription. EMBO J. 18, 2855-2866.
Gall, J. G., and Pardue, M. L. (1969). Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl. Acad. Sci. USA 63, 378-383.
Johnson, C., Primorac, D., McKinstry, M., McNeil, J., Rowe, D., and Lawrence, J. B. (2000). Tracking COLIA1 RNA in osteogenesis imperfecta splice-defective transcripts initiate transport from the gene but are retained within the SC35 domain. J. Cell Biol. 150, 417-432.
Jonh, A., Birnstiel, M. L., and Jones, K. W. (1969). RNA-DNA hybrids at the cytological level. Nature 223, 582-587.
Long, R. M., Elliott, D. J., Stutz, F., Roshbash, M., and Singer, R. H. (1995). Spatial consequences of defective processing of specific yeast mRNAs revealed by fluorescent in situ hybridisation. RNA 1, 1071-1078.
Schwarzacher, T., and Heslop-Harrison, P. (2000). "Practical in Situ Hybridisation." Bios Scientific, London.
Singer, R. H. (1998). Preparation of probes for in situ hybridisation. In "The Singer Lab Online": http://singerlab.aecom.yu.edu/ protocols / insitu_probe_prep.htm.
Wijgerde, M., Grosveld, F., and Fraser, P. (1995). Transcription complex stability and chromatin dynamics in vivo. Nature 377, 209-213.
Xing, Y., Johnson, C. V., Dobner, P. R., and Lawrence, J. B. (1993). Higher level organization of individual gene transcription and RNA splicing. Science 259, 1326-1329.
Xing, Y., Johnson, C. V., Moen, P. T., McNeil, J. A., and Lawrence, J. B. (1995). Non-random gene organization: Structural arrangements of specific pre-mRNA transcription and splicing with SC- 35 domains. J. Cell Biol. 131, 1635-1647.
Zirbel, R. M., Mathieu, U. R., Kurz, A., Cremer, T., and Lichter, P. (1993). Evidence for a nuclear compartment of transcription and splicing located at chromosome domain boundaries. Chrom. Res. 1, 93-106.
Support our developers




