Listeria monocytogenes: Techniques to Analyze Bacterial Infection in vitro
I. INTRODUCTIONListeria monocytogenes is a gram-positive food-borne pathogen responsible for listeriosis, a human infection with an overall 30% mortality rate, ranging from a clinically nonapparent fecal carriage to sever gastroenteritis, mother-to-child infections, and central nervous system infections (Dussurget et al., 2004; Lecuit and Cossart, 2002; Vazquez-Boland et al., 2001). At the individual level, Listeria has the capability to cross three internal barriers: the intestinal barrier, the blood-brain barrier, and the feto-placental barrier. At the cellular level, Listeria is able to enter macrophages, as well as nonprofessional phagocytes, such as epithelial cells. Within infected cells, Listeria disrupts its internalisation vacuole and escapes the phagocytic cascade by proliferating in the cytoplasm. In this environment, Listeria promotes actin polymerisation in order to move inside infected cells and induces its translocation to neighbouring cells, where the intracellular infectious cycle starts again (Fig. 1). Listeria has become a paradigm of bacterial pathogenesis at both cellular and individual levels due to the extensive study of the bacterial virulence factors and of the cellular machinery necessary for the infectious process (Gaillard et al., 1991; Kocks et al., 1992; Dramsi et al., 1995; Ireton et al., 1996; Mengaud et al., 1996; Lecuit et al., 1999; Braun et al., 2000; Shen et al., 2000). This article describes some basic methods in cellular microbiology that have been used routinely to study the infection process of Listeria in vitro that can be adapted to study the pathogenesis of other intracellular bacteria.
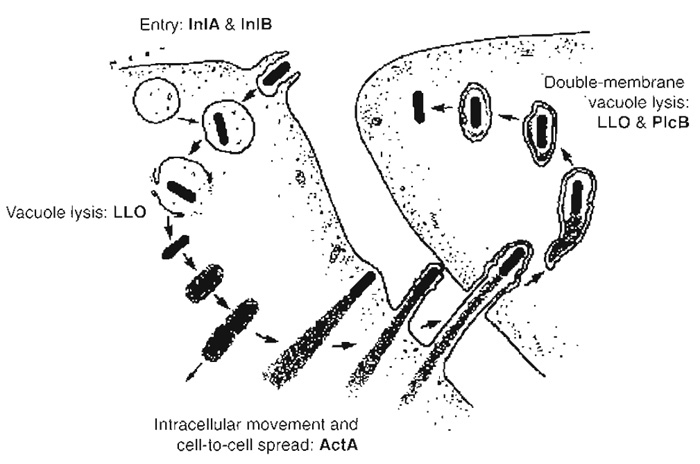 |
| FIGURE 1 Intracellular cycle of Listeria monocytogenes. Listeria induces its own internalisation in nonphagocytic cells through interaction of the bacterial invasion proteins InlA and InlB with their cellular receptors E-cadherin and c-Met, respectively. Bacteria are initially located in a phagocytic vacuole that is disrupted by the lytic enzyme listeriolysin O (LL0), enabling Listeria to reach the cytoplasm and to proliferate in this environment. Intracellular actin-based movement is then induced due to the expression of the bacterial product ActA, which recruits several key players involved in actin polymerisation. Those Listeria that have reached the plasma membrane can form protrusions that extend to neighbouring cells. Bacteria are contained in a double-membrane phagosome that is lysed through the action of listeriolysin O and the phosphatidylcholine phospholipase C PlcB, allowing the infectious cycle to start again. |
II. MATERIALS AND INSTRUMENTATION
Listeria monocytogenes wild-type strain EGD (serovar 1/2a) comes from the bacterial collection in our laboratory at the Pasteur Institute. Lovo cells (Cat. No. CCL-229) are from the American Type Culture Collection. F-12K nutrient mixture (Kaighn's modification, Cat. No. 21127-022), L-glutamine (200mM, 100×, Cat. No. 25030-024), fetal calf serum (Cat. No. 16000-044), trypsin/EDTA (1×, Cat. No. 25300-054), trypan blue (0.4% solution, Cat. No. 15250-061), and phosphatebuffered saline (PBS, pH 7.4, 10×, Cat. No. 70011-036) are from Gibco. Gentamicin (10mg/ml, Cat. No. G- 1272), bovine serum albumin (BSA, Cat. No. A-7030), saponin (Cat. No. S-7900), CaCl2 (Cat. No. C-4901), MgCl2 (Cat. No. M-8266), NaOH (Cat. No. 930-65), Tris (Cat. No. %6066), paraformaldehyde (Cat. No. P-6148), and NH4Cl (Cat. No. A-5666) are from Sigma. Malassez cell counting chamber (Cat. No. 99501.01), glass slides (Cat. No. 79703.01), coverslips (Cat. No. 79720.02), plastic boxes (Cat. No. 96496.02), curved forceps (Cat. No. 24434.01), Whatmann 3 mm chromatography paper (Cat. No. 10343.01), absorbant paper (Cat. No. 62908.01), Parafilm (Cat. No. 97949.01), orbital shaker (Cat. No. 08242.107), beakers (Cat. No. 11362.01), and pH meter (Cat. No. 71519.01) are from Merck-Eurolab. Twenty-four-well plates (Cat. No. 351157), 15-ml polystyrene conical tubes (Cat. No. 352099), petri dishes (Cat. No. 343633), 75-cm2 vented cell culture flasks (Cat. No. 353110), brain-heart infusion agar (Cat. No. 241830), and brain-heart infusion broth (Cat. No. 299070) are from Becton-Dickinson Biosciences. Anti- Listeria rabbit polyclonal serum was developed in our laboratory at the Pasteur Institute (clone R69). The mouse monoclonal anti-E cadherin BTA-1 antibody is from R&D Systems. Rabbit polyclonal antihuman c- Met (Cat. No. C-28) is from Santa Cruz Biotechnology. Goat antirabbit antibodies coupled with Alexa Fluor 488 (Cat. No. A-11008) or Alexa Fluor 546 (Cat. No. A- 11010) and goat antimouse antibodies coupled with Alexa Fluor 488 (Cat. No. A-11001) and Alexa Fluor 488 phalloidin (Cat. No. A-12379) are from Molecular Probes. Mowiol 4-88 (Cat. No. 475904) is from Calbiochem. The tabletop 5415D centrifuge is from Eppendorf. Spectrophotometer Ultrospec 3000Pro is from Amersham Pharmacia Biotech. The axiovert 135 microscope is from Zeiss. Image acquisition is performed with a RTE/CDD-1300 camera by Princeton Scientific Instruments. Image analysis and processing are done with Metamorph software from Universal Imaging Corporation.
III. PROCEDURES
A. Measurement of Bacterial Invasion: The Gentamicin Survival Assay
This procedure is a classical method that can be adapted to study the intracellular survival kinetics of different intracellular pathogens.
Solutions
- Cell culture medium: F-12K nutrient mixture (Kaighn's modification) without supplements. Store at 4°C. Warm at 37°C just before use.
- Complete cell culture medium: F-12K nutrient mixture (Kaighn's modification) supplemented with 2mM L-glutamine and 10% fetal bovine serum. To make 500ml, add 5 ml of a 200mM L-glutamine stock solution and 50ml of fetal bovine serum to 445 ml of F-12K nutrient mixture (Kaighn's modification). Store at 4°C and use within 7 to 8 weeks. Warm at 37°C just before use.
- Complete cell culture medium supplemented with gentamicin: Complete cell culture medium supplemented with 10µg/ml gentamicin. To make 50ml, add 50µl of the gentamicin stock solution (at 10mg/ml) to 50ml of F-12K nutrient mixture. Prepare fresh and warm at 37°C just before use.
- 1× PBS: To make 500ml, add 50ml of the PBS, pH 7.4, 10× solution to 450ml of sterile distilled water. Store at room temperature. Warm at 37°C just before use.
- Trypsin/EDTA: Aliquot in 5-ml portions in sterile 15-ml tubes. Keep at -20°C. Warm at 37°C. just before use.
- Trypan blue solution: Keep at room temperature.
- Brain-heart infusion agar: Suspend 52g of the powder in 1 liter of water and dissolve by frequent agitation. Autoclave at 121°C for 15min, cool down at 50°C, and distribute in petri dishes. Keep at 4°C.
- Brain-heart infusion broth: Suspend 37g of the powder in 1 liter of water and dissolve by frequent agitation. Autoclave at 121°C for 15min. Cool down and keep at room temperature.
Steps
- Two days before the invasion assay, wash the LoVo cells (grown to 90% confluence in a 75-cm2 flask) by removing the cell culture medium and adding 10ml of PBS. Remove the PBS, add 1 ml of trypsin/EDTA, and incubate the cells for 5 min at 37°C in a 10% CO2 atmosphere. After this incubation, carefully hit the flask to release the cells and dilute them in 10ml of fresh complete cell culture medium. Take 10µl of this solution and mix with 90µl of a 0.4% trypan blue solution. With a visible light microscope, count viable cells (the ones not stained by the dye) on a Malassez cell counting chamber (100 squares = 1 µl) and dilute them to a final concentration of 105 cells/ml with complete cell culture medium. Add 500µl of this solution to each of three wells of a 24-well plate and incubate for 48 h at 37°C in a 10% CO2 atmosphere.
- The evening before the invasion assay, take a colony of L. monocytogenes from a brain-heart infusion agar plate and inoculate in 5 ml of brain-heart infusion broth contained in a 15-ml tube. Incubate overnight on a rocking platform at 37°C.
- The day of the assay, dilute 400µl of the bacterial overnight culture in 5 ml of fresh brain-heart infusion broth contained in a 15-ml tube and incubate it on a rocking platform at 37°C to an OD600 of 0.8 (approximately 8 × 108 bacteria/ml).
- Centrifuge 1 ml of the bacterial culture for 2min at 6000rpm and resuspend the pellet in 1ml of PBS. Repeat this step twice; the second time resuspend the bacterial pellet in 1 ml of cell culture medium. Measure the OD and dilute bacteria to a final solution of 107 bacteria/ ml with cell culture medium.
- Wash LoVo cells in each well once with 1ml of cell culture medium and add 500µl of the bacterial dilution (MOI of 50, as we assume that cells have grown to a final density of 105 cells/well). Incubate for 1h at 37°C in a 10% CO2 atmosphere.
- Make a 10-fold dilution of the initial bacterial solution by adding 100µl of this preparation to 900µl of sterile distilled water contained in an Eppendorf tube. Make another 10-fold dilution by adding 100µl of the first 10-fold dilution to 900µl of sterile distilled water contained in a different Eppendorf tube. Repeat this operation twice. Take a brain-heart infusion agar plate, divide the surface into four equal parts (by tracing a cross on the bottom of the plate), and plate 50 µl of each 10-fold dilution on each part. Incubate the plate overnight at 37°C in order to have an exact record of the precise number of bacteria that were inoculated on LoVo cells.
- After 1 h of bacterial inoculation, wash LoVo cells carefully three times with 1ml of PBS and then add 1 ml of the cell culture medium supplemented with gentamicin. Incubate the cells for another hour at 37°C in a 10% CO2 atmosphere.
- Wash the cells twice with 1 ml of PBS and add 1 ml of sterile distilled water to each well. Disrupt the cells by pipetting up and down water several times. Make a 10-fold dilution by adding 100µl of the disrupted cell solution contained in each well to a different Eppendorf containing 900µl of steriled distilled water. Make another 10-fold dilution for each tube by adding 100 µl of the first 10-fold dilution to 900µl of sterile distilled water contained in a different Eppendorf tube. Repeat this operation once for each individual series of tubes. Take three brain-heart infusion agar plates, divide their surface in four equal parts (by tracing a cross on the bottom of the plate), and for each series of tubes, plate 50µl of each 10-fold dilution on a different part of the agar plate (also plate 50 µl of the initial disrupted cell solution contained on the wells). Incubate the plate overnight at 37°C.
- The day after the invasion assay, count the colony-forming units (CFU) on the agar plates at a dilution that allows a clear discrimination of individual bacterial colonies. The percentage of Listeria that were able to invade the LoVo cells (and which survived the gentamicin treatment) is evaluated as the ratio of bacteria recovered from the disrupted cell solution divided by the total number of bacteria added to the monolayers (results are expressed as the mean of bacterial counts obtained from the three different wells ± standard deviation).
Comments
- Cells are grown preferentially without antibiotics.
- Listeria are P2 microorganisms and should be manipulated in a microbiological hood.
- The replication of bacteria inside cells can be analysed over time if different subsets of cells are infected initially at the same time and then each subset of cells is lysed at different time points after inoculation. This procedure is helpful in order to track the proliferation or killing of bacteria in the intracellular environment.
B. Measurement of Bacterial Invasion: Differential Immunofluorescence Labeling of Intracellular versus Extracellular Bacteria
Visualisation of bacteria allows evaluation of several parameters, such as the proportion of cells in the inoculated monolayer to which bacteria attach, as well as the proportion of adherent versus invasive bacteria.
Solutions
- Cell culture medium, complete cell culture medium, 1× PBS, trypsin/EDTA, trypan blue solution, brain-heart infusion agar, and brain-heart infusion broth: Prepare as described in the Section III,A.
- Fixation solution: 3% paraformaldehyde in PBS. To prepare 100ml, heat 80ml of PBS to 80°C and then add 3 g of paraformaldehyde while stirring. Mix until clear (add a few drops of 1N NaOH to help dissolution if the liquid does not become clear after several minutes of stirring). Add 100µl of 100mM CaCl2 and 100µl of 100mM MgCl2, cool down at room temperature, add PBS up to 100ml, and check pH (should be about 7.4). Aliquot in 5-ml portions and store at -20°C. Use only freshly thawed solution.
- Quenching solution: 50 mM NH4Cl in PBS. Prepare 100ml of a stock solution of 1M NH4Cl in PBS by adding 5.35g of NH4Cl to 80ml of PBS. Mix until clear and add PBS up to 100ml. Keep at room temperature. The quenching solution used in the experiment will be prepared freshly by diluting 500µl of the 1M NH4Cl stock solution in 9.5 ml of PBS.
- Blocking solution: 1% BSA in PBS. To prepare 200 ml, add 2 g of BSA to 200 ml of PBS. Mix until clear. Prepare fresh just before performing the experiment.
- Permeabilising/blocking solution: 0.05% saponin in blocking solution (1% BSA in PBS). To prepare 100ml, add 50µg of saponin to 100ml of blocking solution. Prepare fresh just before performing the experiment.
- First primary antibody solution: 1/500 dilution of R11 anti-Listeria rabbit polyclonal serum in blocking solution. To prepare 500µl, add 1 µl of the R11 serum in 499µl of blocking solution (1% BSA in PBS). Prepare fresh before performing the experiment.
- First secondary antibody solution: 1/500 dilution of Alexa 546 antirabbit serum in blocking solution. Prepare as in step 6.
- Second primary antibody solution: 1/500 dilution of R11 anti-Listeria rabbit polyclonal serum in permeabilising solution. Prepare as in step 6, using permeabilising/ blocking solution instead of blocking solution.
- Second secondary antibody solution: 1/500 dilution of Alexa 488 antirabbit serum in permeabilising solution. Prepare as in step 7, using permeabilising/blocking solution instead of blocking solution.
- Mounting solution: 2.4% Mowiol/6% glycerol in 20mM Tris buffer. To prepare 100ml, add 2.4g of Mowiol and 6g of glycerol to 6 ml of distilled water. Keep at room temperature overnight. Add 90ml of 0.2M Tris, pH 8.5, warm in a water bath at 50°C, and mix regularly until total dissolution. Spin 15min at 4000rpm and aliquot supernatant in 1-ml portions at -20°C. Once an aliquot is thawed, keep at 4°C.
Steps
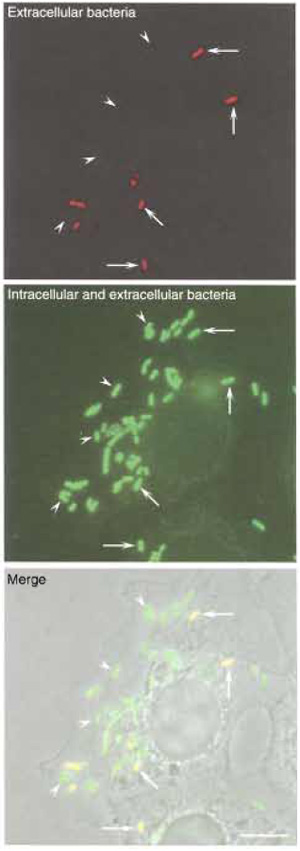 |
| FIGURE 2 Differential immunolabeling of intracellular versus extracellular bacteria. LoVo cells were infected with L. monocytogenes as described in the text, and extracellular bacteria were distinguished from intracellular microorganisms by differential immunolabeling. (Top) Extracellular bacteria were labeled with an anti-Listeria serum without permeabilising LoVo cells. (Middle) The total population of bacteria (both extracellular and intracellular) was labeled with the same anti-Listeria serum after permeabilising LoVo cells. Intracellular bacteria are identified as those bacteria that are labeled only after the permeabilisation treatment. (Bottom) Phase image of LoVo cells and Listeria. Bar: 5 µm. Arrowheads, intracellular bacteria; arrows; extracellular bacteria. |
- Infect LoVo cells following steps 1 to 5 described in Section III,A, adding 12-mm-diameter glass coverslips to the wells before seeding the cells (step 1).
- After 1 h of bacterial inoculation, wash LoVo cells carefully three times with 1ml of PBS and then add 500µl of the fixation solution. Incubate for 15 min (all steps are now performed at room temperature).
- Wash LoVo cells carefully once with 1 ml of PBS and add 500µl of the quenching solution in order to block free aldehyde groups. Incubate for 10min.
- Prepare a wet incubation chamber by humidifying a 10 × 10-cm piece of Whatman 3mm chromatographic paper. Add a sheet of Parafilm over the Whatman paper and, for each coverslip that is going to be labelled, add a drop of 30µl of the first primary antibody solution to the Parafilm sheet.
- Take carefully each coverslip from the wells with the forceps, carefully remove the excess of liquid from the coverslip by touching gently the tip of the coverslip with absorbant paper, and label extracellular bacteria by putting the coverslip (cells downwards) over the drop of the first primary antibody solution. Cover with a plastic lid and incubate for 30min.
- Prepare a second wet incubation chamber as described in step 4 and add a drop of 30µl of the first secondary antibody solution to the Parafilm sheet for each coverslip that is going to be labeled.
- Take carefully each coverslip with the forceps and, in order to wash the first primary antibody solution, dip it gently 10 times in a 50-ml beaker containing 40ml of the blocking solution. Remove excess liquid as described in step 5 and label the first primary antibody by putting the coverslip (cells downwards) over the drop of the first secondary antibody solution. Cover with the plastic lid and incubate for 25 min.
- Prepare a third wet incubation chamber as described in step 4 and, for each coverslip that is going to be labelled, add a drop of 30µl of the second primary antibody solution to the Parafilm sheet.
- Take carefully each coverslip with the forceps and wash the first secondary antibody solution by dipping the coverslip 10 times in a 50-ml beaker containing 40ml of the blocking solution and then dipping it again 10 times in a 50-ml beaker containing 40ml of the permeabilising/blocking solution. Remove excess liquid from the coverslip as described in step 5 and label intracellular and extracellular bacteria by putting the coverslip (cells downwards) over the drop of the second primary antibody solution. Cover with the plastic lid and incubate for 30min.
- Prepare a fourth wet incubation chamber as described in step 5 and add a drop of 30µl of the second secondary antibody solution to the Parafilm sheet for every coverslip that is going to be labelled.
- Take carefully each coverslip with the forceps and wash the second primary antibody solution by dipping the coverslip 10 times in a 50-ml beaker containing 40ml of the permeabilising/blocking solution. Remove excess liquid as described in step 5 and label the second primary antibody by putting the coverslip (cells downwards) over the drop of the second secondary antibody solution. Cover with the plastic lid and incubate for 25 min.
- Take carefully each coverslip and wash the second secondary antibody solution by dipping it 10 times in a 50-ml beaker containing 40ml of the permeabilising/ blocking solution, 10 times in a different 50-ml beaker containing 40 ml of PBS, and 10 times in a third 50-ml beaker containing 40 ml of distilled water. Remove excess water and put the coverslip carefully over a 10-µl drop of mounting medium on top of a glass slide.
- Visualise the preparation with a fluorescence microscope (Fig. 2). Extracellular bacteria, labelled with the first and second primary antibodies and, accordingly, also labelled with the first and second secondary antibodies, will appear yellow (due to the superposition of the green signal of the Alexa 488 fluorochrome and of the red signal of the Alexa 546 fluorochrome). Intracellular bacteria, however, labelled only with the second primary antibody and, consequently, labelled only with the second secondary antibody, will appear green. Count extracellular and intracellular Listeria in at least 100 LoVo cells and express the results in a histogram standardising the total number of bacteria to 100% and expressing accordingly the corresponding proportions of extracellular and intracellular Listeria.
Comments
- Never allow cells to dry.
- As fluorochromes are sensitive to light, perform incubations in the dark by covering with a plastic lid enveloped in aluminum foil.
- Antibody dilutions are suggested accordingly to their use in our own laboratory. They could need finer adjustment depending on the batch used.
C. Visualisation of the Interaction of Listeria with Its Receptors E-Cadherin and c-Met
Two bacterial proteins, InlA (internalin) and InlB, are known to mediate the internalisation process of Listeria into target cells (Gaillard et al., 1991; Dramsi et al., 1995; Cossart et al., 2003). InlA promotes entry into a subset of epithelial cells that express its cellular receptor, the adhesion molecule E-cadherin (Mengaud et al., 1996; Lecuit et al., 1999), a transmembrane protein that is normally involved in homophilic cell/cell interactions. Two different cellular receptors have been identified for InlB: the receptor for the globular part of the complement component C1q (gC1q-R) (Braun et al., 2000) and c-Met, the receptor for the hepatocyte growth factor (Shen et al., 2000). Interaction of InlA and InlB with their respective cellular receptors induces the recruitment to the site of bacterial entry of intracellular molecules that mediate cytoskeletal rearrangements necessary to induce bacterial invasion (Ireton et al., 1996, 1999; Lecuit et al., 2000; Bierne et al., 2001). By immunofluorescence it is possible to visualise the recruitment of E-cadherin and c-Met at the site of bacterial invasion.
Solutions
- Cell culture medium, complete cell culture medium, 1× PBS, trypsin/EDTA, trypan blue solution, brain-heart infusion agar, and brain-heart infusion broth: Prepare as described in Section III, A.
- Fixation solution, quenching solution, permeabilising/ blocking solution, and mounting medium: Prepare as described in Section III,B.
- Primary antibody solution: 1/100 dilution of both the anti-E-cadherin mouse monoclonal serum BTA-1 and the anti-C-Met polyclonal serum C-28. To prepare 500 µl, add 5 µl of the anti-E-cadherin BTA-1 serum and 5 µl of the anti-C-Met C-28 serum to 490µl of permeabilising/ blocking solution. Prepare fresh before performing the experiment.
- Secondary antibody solution: 1/500 dilution of both Alexa 488 antimouse serum and Alexa 546 antirabbit serum in permeabilising/blocking solution. To prepare 500 µl, add 1 µl of each secondary antibody to 498 µl of permeabilising/blocking solution freshly before use.
Steps
- Infect LoVo cells following step 1 in Section III,B.
- Fix and quench LoVo cells following steps 2 and 3 in Section III,B.
- Label LoVo cells following steps 4 to 7 in Section III,B, taking into account that in the present experiment there is only one primary antibody solution (containing actually a mix of two different primary antibodies) and only one secondary antibody solution (containing a mix of two different secondary antibodies).
- Wash and mount the cells following step 12 in Section III,B.
- Visualise the preparation with a fluorescence microscope (Fig. 3). c-Met will be recognised by the red signal (from the Alexa 546 fluorochrome) and will be observed at different locations of the plasma membrane of LoVo cells. E-cadherin will be detected by the green signal (from the Alexa 488 fluorochrome) and will be found essentially at adherens junctions located in the borderline region between two different adjacent cells. Listeria can be visualised by light microscopy (phase contrast). Both receptors can also be detected at the site of entry of invading Listeria.
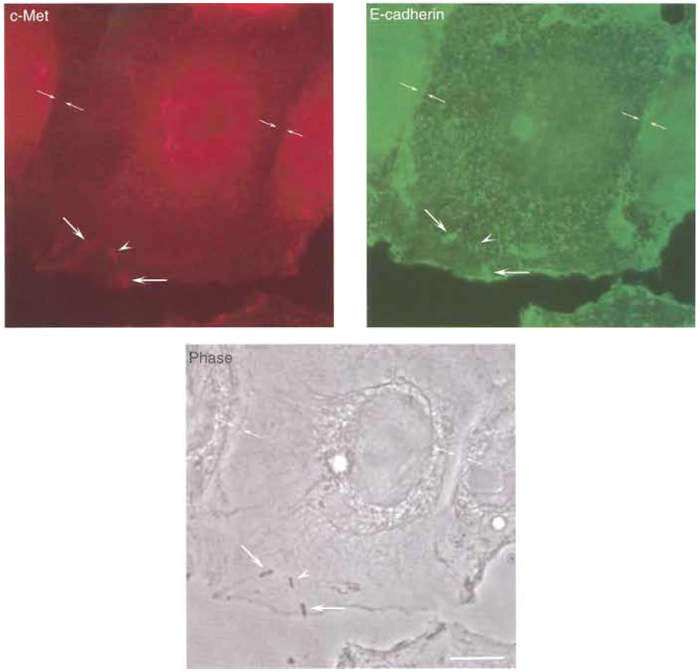 |
| FIGURE 3 Interaction of Listeria with its receptors E-cadherin and c-Met. LoVo cells were infected with L. monocytogenes as described in the text, and its receptors E-cadherin and c-Met were immunolabelled. (Top left) c-Met shows a diffuse distribution in LoVo cells and is clearly absent at sites of adherens junctions formation, but is clearly recruited at sites of bacterial invasion. While some of the invading Listeria colocalise with both E-cadherin and c-Met, one bacterium is shown that only colocalises with c-Met and not with Ecadherin (arrowhead), suggesting that the interaction of Listeria with each one of their receptors can be uncoupled, as observed in cells lines such as Caco-2 (in which invasion is mediated mainly by the InlA/E-cadherin interaction) or Veto (in which invasion is driven exclusively by the InlB/c-Met interaction). (Top right) Ecadherin distribution at the plasma membrane of LoVo cells can be clearly seen at sites of adherens junction formation as well as at sites of bacterial entry. (Bottom) Phase image of LoVo cells and Listeria. Bar: 5µm. Opposing arrows, adherens junction; large arrows, bacteria that recruit E-cadherin and c-Met at their site of entry; arrowheads, bacterium that only recruits c-Met at its site of entry. |
D. Visualisation of Listeria.Induced Actin Comet Tails and Protrusions
A hallmark of Listeria infection is the polymerisation of cellular actin: bacteria that have escaped their internalisation vacuoles take advantage of this actin-based motility mechanism to reach and infect neighbouring cells without leaving the intracellular environment. A single bacterial product, the protein ActA, is responsible for this phenomenon by recruiting to the bacterial rear end two cellular key players of the actin nucleation process: VASP and the Arp2/3 complex (Cossart, 2000). Polymerised actin can be visualised as a structure that extends from the rear end of the moving Listeria to the cell cytoplasm, which has been named the actin comet tail. Bacteria that have reached the plasma membrane of a primary infected cell form protrusions that extend into the cytoplasm of neighbouring cells (or that extend simply to the extracellular space in the case of cells that do not have adjacent neighbours). These structures can be visualised by labelling the cellular actin with the fungal drug phalloidin in cells that have been infected with Listeria for at least 5 h.
Solutions
- Cell culture medium, complete cell culture medium, 1× PBS, trypsin/EDTA, trypan blue solution, brain-heart infusion agar, and brain-heart infusion broth: Prepare as described in Section III,A.
- Fixation solution, quenching solution, permeabilising/ blocking solution, and mounting medium: Prepare as described in Section III, B.
- Primary antibody solution: 1/500 dilution of R11 anti-Listeria rabbit polyclonal serum in permeabilising/ blocking solution. To prepare 500µl, add 1µl of the R11 serum in 499µl of permeabilising/blocking solution. Prepare fresh before performing the experiment.
- Secondary antibody and probe solution: 1/500 dilution of Alexa 546 antirabbit serum in permeabilising/ blocking solution and 1/100 dilution of phalloidin Alexa 488. To prepare 500µl, add 1µl of the Alexa 546 serum and 5µl of the phalloidin Alexa 488 probe to 496 µl of permeabilising/blocking solution. Prepare freshly before use.
Steps
- Infect LoVo cells following steps 1 to 5 and step 7 in Section III,A, adding 12-mm-diameter glass coverslips to the wells before seeding the cells (step 1) and incubating the LoVo cells in the presence of cell culture medium supplemented with gentamicin for 5 h insted of 1h (step 7).
- Fix and quench LoVo cells following steps 2 and 3 in Section III,B.
- Label LoVo cells following steps 4 to 7 in Section III,B, taking into account that in the present experiment there is only one primary antibody solution (containing one primary antibody) and only one secondary antibody solution (containing a mix of one secondary antibody and one cytoskeletal probe).
- Wash and mount the cells following step 12 in Section III,B.
- Visualise the preparation using a fluorescence microscope (Fig. 4). Actin will be detected by the green signal (from the Alexa 488 fluorochrome) and will be observed at different cellular locations, such as stress fibres or cortical filaments near the plasma membrane. Listeria will appear as red (due to the signal of the Alexa 546 fluorochrome). Bacteria that have just lysed their phagosomal membrane and that have already started to polymerise actin will also be surrounded uniformly with the Alexa 488 fluorochrome. Those Listeria that broke their vacuole early will be associated with comet tails, which can be visualised starting at the rear pole of red bacteria and projecting into the cell cytoplasm. In the case of those Listeria that have reached the plasma membrane of an infected cell, a protrusion can be seen as a protruding structure that contains a red bacteria at its tip and a green actin comet tail following behind.
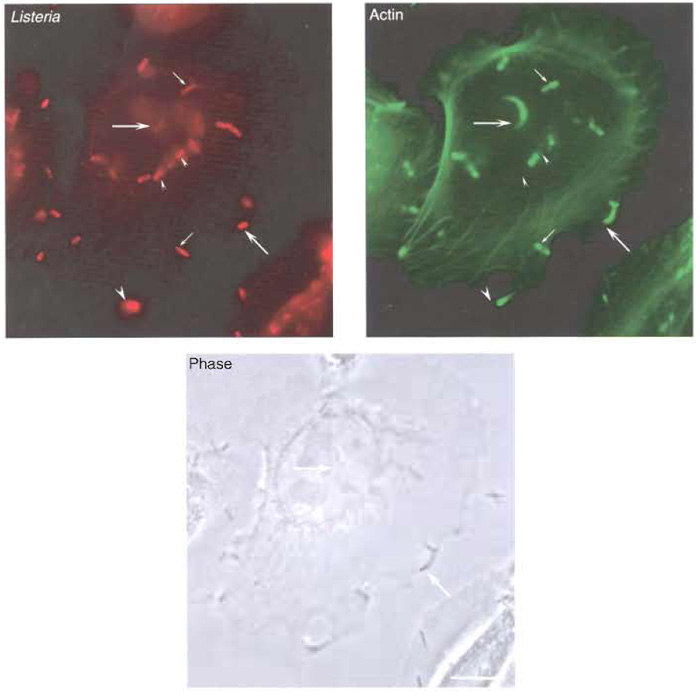 |
| FIGURE 4 Actin polymerisation and protrusion formation by Listeria. LoVo cells were infected with Listeria as described in the text. Both bacteria and the actin cytoskeleton were labelled for immunofluorescence analysis. (Top left) Labelling of the total population of bacteria associated to LoVo cells. (Top right) Actin labelling with the fungal drug phalloidin. While some bacteria are not associated with actin (small arrowheads), other bacteria are detected at early phases of actin polymerisation (small arrows). Bacteria at later stages of their intracellular cycle can be observed at the tip of actin comet tails (big arrows). Listeria that have reached the plasma membrane can form protrusions that extend beyond the infected cell (big arrowhead). (Bottom) Phase image of LoVo cells and Listeria. Bar: 5 µm. |
References
Bierne, H., Gouin, E., Roux, E, Caroui, E, Yin, H.L., and Cossart, E (2001). A role for cofilin and LIM Kinase in Listeria induced phagocytosis. J. Cell Biol. 155, 101-112.
Braun, L., Gebrehiwee, B., and Cossart, E (2000). gC1q-R/p32, a C1q binding protein, is a novel receptor for Listeria monocytogenes. EMBO I. 19, 1458-1466.
Cossart, E (2000). Actin-based motility of pathogens: The Arp2/3 complex is a central player. Cell. Microbiot. 2, 195-205.
Cossare, P., Pizarro-Cerda, J., and Lecuit, M. (2003). Invasion of mammalian cells by Liseeria monocyeogenes: Functional mimicry to subvert cellular functions. Trends Cell Biol. 13, 23-31.
Dramsi, S., Biswas, I., Maguin, E., Braun, L., Mastroeni, E, and Cossare, E (1995). Entry of L. monocytogenes into hepatocytes requires expression of InlB, a surface protein of the internalin multigene family. Mol. Microbiol. 16, 281-261.
Dussurgee, O., Pizarro-Cerda, J., and Cossart, P. (2004). Molecular determinauts of Listeria monocytogeues virulence. Annu. Rev. Microbiol. 58, 587-610.
Galliard, J.-L., Berche, E, Frehel, C., Gouin, E., and Cossart, E (1991). Entry of L. monocytogenes into cells is mediated by ineernalin, a repeat protein reminiscent of surface antigens from Grampositive cocci. Cell 65, 1127-1141.
Ireton, K., Payrastre, B., and Cossart, E (1999). The Listeria monocytogenes protein InlB is an agouist of manmalian phosphoinositide 3-kinase. J. Biol. Chem. 274, 17025-17032.
Ireton, K., Payrastre, B., Chap, H., Ogawa, W., Sakaue, H., Kasuga, M., and Cossart, P. (1996). A role for phosphoinositide 3-kinase in bacterial invasion. Science 274, 780-782.
Kocks, C., Gouin, E., Tabouret, M., Berche, P., Ohayon, H., and Cossart, P. (1992). L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68, 521-531.
Lecuit, M., and Cossart, P. (2002). Genetically-modified animal models for human infections: The Listeria paradigm. Trends Mol. Med. 8, 537-542.
Lecuit, M., Dramsi, S., Gottardi, C., Fedor-Chaiken, M., Gumbiner, B., and Cossart, P. (1999). A single amino-acid in E-cadherin is responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J. 18, 3956-3963.
Lecuit, M., Hurme, R., Pizaro-Cerda, J., Ohayon, H., Geiger, B., and Cossart, P. (2000). A role for alpha- and beta-catenins in bacterial uptake. Proc. Natl. Acad. Sci. USA 97, 10008-10013.
Mengaud, J., Ohayon, H., Gounon, P., M6ge, J.-M., and Cossart, P. (1996). E-cadherin is the receptor for internalin, a surface protein required for entry of Listeria monocytogenes into epithelial cells. Cell 84, 923-932.
Shen, Y., Naujokas, M., Park, M., and Ireton, K. (2000). InlBdependent internalization of Listeria is mediated by the met receptor tyrosine kinase. Cell 103, 501-510.
Vazquez-Boland, J.A., Kuhn, M., Berche, P., Chakraborty, T., Dominguez-Bernal, G., Goebel, W., Gonzalez-Zorn, B., Wehland, J., and Kreft, J. (2001). Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14, 584-640.
Support our developers




