Production and Quality Control of High-Capacity Adenoviral Vectors
I. INTRODUCTIONHigh-capacity adenovirus (HC-Ad) vectors [also called pseudoadenovirus (PAV), helper-dependent (HD-Ad), gutted, or gutless adenovirus vectors] have been developed to address capacity, toxicity, and immunogenicity problems of first- and secondgeneration adenovirus vectors. As only viral elements this vector type contains the inverted terminal repeats (ITRs), which are essential for replication of the viral DNA, and the packaging signal close to the left terminus that is required for encapsidation of the DNA into the viral capsids. Since the size of the ITRs and the packaging signal together are less than 0.6kb, up to 37kb of foreign DNA can be transported.
For practical reasons, most HC-Ad vectors will carry genes or expression cassettes that are smaller than 37kb. For stability reasons during amplification in most cases additional "stuffer" DNA has to be incorporated into the vector DNA to increase the genome size to at least around 27kb.
HC-Ad vectors cannot be produced similar to helper-independent vectors, in which most viral functions are provided from the vector. Because adenovirus is a relatively large DNA virus that expresses many different protein and RNA functions, it is unlikely that complementing cell lines can be generated to provide appropriate levels of all viral functions in trans. Therefore, a helper virus is used for production that subsequently is eliminated from the end product.
The currently preferred production system is based on excision of the packaging signal of the helper virus by a recombinase expressed in the producer cell line. Most HC-Ad vectors so far have been produced using the Cre-loxP recombination system of the bacteriophage PI. In this system the packaging signal of the helper virus is flanked by two loxP sites (Hardy et al., 1997; Parks et al., 1996). The HC-Ad vector is produced in El-complementing cells that express the recombinase constitutively. The Cre-mediated excision is surprisingly efficient and the contamination of vector by helper virus is reduced compared to the earlier production system.
The complete characterization of HC-Ad vector preparations comprises three parameters: (1) the number of infectious particles, (2) the number of total particles, and (3) the number helper virus particles remaining in the preparation after purification (Kreppel et al., 2002). Due to the fact that HC-Ad vectors do not possess any viral coding sequences, the number of infectious particles cannot be determined by plaque assay or tissue culture infectivity dose TCID50 because these methods rely on vector replication and viral protein expression in El-transformed cell lines. The number of total particles can be determined by particle lysis and subsequent measuring light absorbance at 260nm. However, the reliability of this method is usually low and strongly depends on the purity of the vector preparations. The DNA-based method described here allows for fast and reliable determination of all three parameters with standard laboratory equipment independent of viral or reporter gene expression. For quantifying the number of infectious particles, a reference cell line with defined susceptibility for Ad5 is transduced with the HC-Ad vector, cell lysates are prepared, and vector genomes that entered the cells are detected after immobilization on a nylon membran by hybridization with a radiolabeled vector-specific probe. Total particle numbers can easily be determined with the same probe on the same membrane by preparing particle lysates and immobilizing the DNA. Finally, by choosing a probe specific for the helper virus genome the number of helper virus particles can be quantified in the particle lysates.
This article is divided in two parts: in the first part the production process includes serial amplification in producer cells and ends with CsCl density centrifugation for purification. In the second part a method for titration of purified vectors is described that is based on slot blot methods and that not only allows one to determine particle and infectious vector titers, but also the contamination with helper virus.
II. PRODUCTION OF HC-Ad VECTORS
Materials
Cells and Plasmids
El-Expressing Cell Lines
Several cell lines have been described that express the adenovirus type 5 (Ad5) early region E1 (E1A and EI B) and thus support the growth of El-deleted vectors and helper viruses (Fallaux et al., 1998; Gao et al., 2000; Graham et al., 1977; Schiedner et al., 2000). In contrast to HEK293 cells, PER.C6 and N52.E6 cells have been designed not to produce replicationcompetent adenovirus (RCA) and thus are a preferred cell type for the production of El-deleted vectors and helper viruses.
Cre-Expressing Cell Lines
The production process of HC-Ad vectors builds on two viral components; vector and El-deleted helper virus with packaging signal flanked by loxP sites and serial amplifications in producer cells expressing Crerecombinase. Excision of the packaging signal from the helper virus in the producer cells results in preferential packaging of vector genomes and a reduction of helper virus contamination. Several HEK293-based Cre-expressing cell lines have been described (Hilgenberg et al., 2001; Parks et al., 1996). Similar to first-generation adenoviral vectors, the helper virus is prone to turn into RCA when HEK-293 based cells are used for HC-Ad vector production. Based on PER.C6, a Cre-expressing cell line has been described preventing the occurrence of RCA in HC-Ad vector preparations (Sakhuja et al., 2003). Similarly, a Cre-expressing cell line 73/29 has been generated (manuscript submitted) that is based on N52.E6 cells.
Helper Virus Plasmids
A preferred helper virus used for preparation of HC-Ad vectors contains loxP (Parks et al., 1996) or frt (Ng et al., 2001; Umana et al., 2001) sites flanking the packaging signal. Consequently, in Cre- or FLPerecombinase expressing producer cells the packaging signal of the helper virus is excised and vector genomes are preferentially packaged. Most published helper viruses are E1 and E3 deleted and contain a reporter gene cassette in E3 in order to allow easy quantitation of helper virus contamination (Hartigan- O'Connor et al., 2002; Hilgenberg et al., 2001; Ng et al., 2001; Palmer and Ng, 2003; Parks et al., 1996; Sandig et al., 2000; Umana et al., 2001). However, this insertion may affect helper virus yield and can provoke an immune reaction against the reporter in vivo. Recombination events between helper and vector inverted terminal repeats (ITRs) occur frequently and can result in loss of a loxP site in the helper virus genomes and thus outgrowth of mutated helper virus. Figure 1 shows the schematic structure of helper virus plasmid pGS102#21, which contains Ad5 sequences from nucleotides 1-341 with a loxP site introduced at nucleotide 192, a second loxP site introduced at nucleotide 341 followed by 4.6 kb of λ DNA, and Ad5 sequence nucleotides 3523 to 35935. The adenoviral packaging signal consists of seven functional units called A repeats with the consensus sequence [ATTTGN8GC] (Schmid and Hearing, 1998). In order to minimize sequence homologies between vector and helper virus genome, pGS102#21 contains only A repeats I to IV. In addition, sequences from phage λ were inserted in the helper virus genome in order to increase the size of the helper virus, thus improving separation of helper and vector particles in a CsCl- gradient, pGS102#21 is an infectious plasmid based on pBluescript with unique SwaI sites flanking both ITRs.
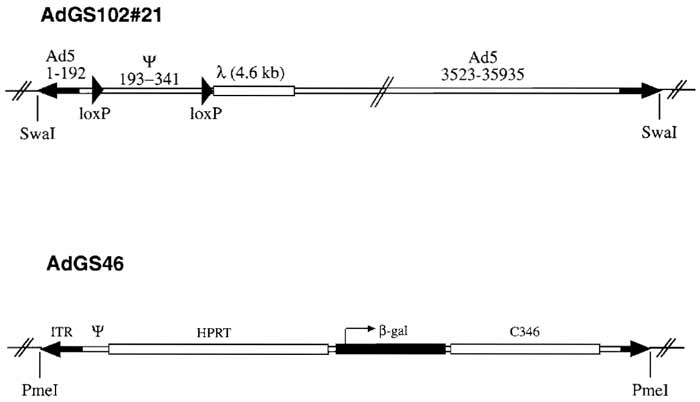 |
| FIGURE 1 Structure of helper virus AdGS102#21 and HC-Ad vector AdGS46. Helper virus AdGS102#21 is E1 deleted and contains Ad5 sequences from nucleotides 1 to 341 with a loxP oligo nucleotide introduced at nucleotide 192, a second loxP oligo nucleotide introduced at nucleotide 341 followed by 4.6 kb of λ, DNA, and Ad5 sequences from nucleotides 3523 to 35935. AdGS46 contains the Ad5 left terminus, 16 kb of HPRT stuffer, a β-galactosidase expression cassette, 9kb of C346 stuffer DNA, and the Ad5 right terminus. Both AdGS102#21 and AdGS46 were constructed as plasmids based on pBluescript with unique SwaI (AdGS102#21) and PmeI (AdGS46) sites flanking both inverted terminals for easy release of the plasmid backbone. |
Plasmids for Cloning HC-Ad Vectors
Early observations using different sizes of HC-Ad vectors suggested that only vector genomes with sizes of at least 27kb allowed efficient and stable vector amplification (Parks and Graham, 1997). The sizes of most expression cassettes used currently for gene transfer are smaller than this 27-kb minimal size for HC-Ad vectors. Therefore, additional DNA has to be incorporated into the vector genome as stuffer DNA. Since the source of the stuffer sequences may influence transgene expression (Parks et al., 1999), the use of noncoding DNA from human origin is preferred. Figure 2 shows different plasmids constructed for the incorporation of different sized transgenes. Most plasmids contain noncoding stuffer sequences derived either from the human HPRT locus or from the human cosmid C346. In addition, all plasmids contain left (nucleotides 1-440) and right (nucleotides 35818-35935) termini of Ad5 DNA. Both left and right adenoviral termini are flanked by unique PmeI or SnaBI (pSTK142) restriction sites. The plasmid backbone in all constructs is pBluescript.
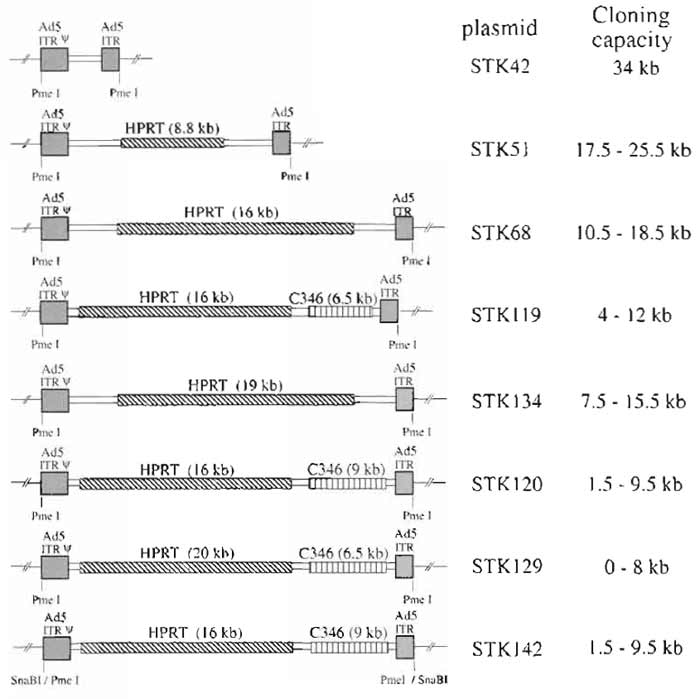 |
| FIGURE 2 Plasmids for cloning HC-Ad vectors. Plasmids contain Ad5 left (Ad5 sequences nucleotides 1 to 440) and right (Ad5 sequences nucleotides 35818 to 35935) termini. In addition, cloning plasmids contain noncoding stuffer sequences from the HPRT gene locus or the C346 cosmid. Again, to release the pBluescript backbone, both inverted terminal repeats are flanked by PmeI sites (or SnaBI for pSTK42). In order to incorporate different sizes of transgenes, the plasmids contain several unique cloning sites. |
The HC-Ad vector AdGS46 has been described previously (Thomas et al., 2000) and is based on pSTK120. Plasmid pGS46 contains the Ad5 left terminus, 16 kb of HPRT stuffer, a β-Gal expression cassette containing the hCMV promoter and SV40 polyadenylation signal, a 9-kb stuffer from C346, and the Ad5 right terminus (Fig. 1).
Cell Culture Reagents
All tissue culture reagents, including (x-modified Eagle's medium (αMEM, Cat. No. 12000-063), fetal calf serum (FBS, Cat. No. 10270-106), phosphate-buffered saline (PBS, Cat. No. 14190-169), trypsin solution (Cat. No. 25300-096), and antibiotics (Cat. No. 10378-016) are from Invitrogen. G418 for selection of neomycin expression is from Sigma (geneticin, Cat. No. G5013). Tissue culture dishes (6- and 15-cm dishes) are from Renner.
Additional Reagents and Solutions
Centrifuge tubes: SW41 ultraclear centrifuge tubes (Beckmann, Cat. No. 344059)
Desalting column: PD-10 column (Amersham Bioscience, Cat. No. 17-0851-01)
Chloroform/isoamyl alcohol: Mix 10ml isoamyl alcohol with 230ml chloroform
TE: 10 mM Tris, 1 mM EDTA, pH 7.5
0.5M EDTA: Dissolve 186.1 g EDTA-2H2O in 800ml H2O, adjust pH to 8.0 using NaOH pellets (approximately 20 g), fill up to 1 liter, and autoclave
MEM agarose overlay: Dissolve 1g agarose (AppliChem, Cat. No. A2114,0500) in 100ml H2O, autoclave, and cool to 40°C. Add prewarmed (37°C) 90ml 2× MEM (Gibco, Cat. No. 61100-087), 10ml FBS, 1 ml antibiotics (Gibco, Cat. No. 10378-016), and 2 ml 5% yeast extract
Na-acetate (3M): Dissolve 24.6g Na-acetate in 100ml H2O, adjust pH to 5.2 using acetic acid, and autoclave
Phenol, buffer saturated: Gibco (Cat. No. 15513-039)
Proteinase K (Sigma, Cat. No. P6556): 5mg/ml dissolved in sterile H2O
QIAamp DNA Minikit: Qiagen (Cat. No. 51104)
RNase: DNase free (Roche, Cat. No. 1119915)
10% SDS: Dissolve 10 g SDS in 100 ml sterile H2O
Superfect transfection kit (Qiagen Cat. No. 301305)
Tris-buffered saline (TBS): 25 mM Tris, 137 mM NaCl, 2.7 mM KCl, pH 7.4, autoclave
TBS/10% glycerol: TBS containing 10% glycerol
TBS/CsCl: Dissolve 10g CsCl (Roche, Cat. No. 757306) in 20 ml sterile TBS
Ultracentrifuge, SW41 rotor: e.g., Beckmann
Yeast extract (5%): Dissolve 2g yeast extract (AppliChem, Cat. No. A1552,0100) in 40ml H2O, sterile filter
Production of Helper Virus
Preparation of DNA for Transfection
- Digest 10µg of plasmid pGS102#21 with 20-50 units of SwaI (New England Biolabs) at 25°C for 2h.
- Add TE buffer, pH 8.5, up to 200µl, add 200µl phenol, vortex gently, and precipitate for 5min at 14,000 rpm.
- Take upper phase, add 200µl chloroform/ Isoamyl alcohol, vortex gently, and centrifuge for 5 min at 14,000 rpm.
- Take upper phase, add 20 µl 3 M Na-acetate, mix, add 400µl ethanol, mix, and precipitate for 30min at 14,000rpm and 4°C.
- Discard supernatant, add 400µl 70% ethanol, centrifuge for 10 min at 14,000 rpm and 4°C. and discard supernatant.
- Dissolve pellet in 20µl TE, pH 7.5 (0.5µg/µl).
Preparation of N52.E6 Cells for Transfection
N52.E6 cells are used for the production of helper virus and are cultivated in αMEM supplemented with 10% FBS and antibiotics in 5% CO2 at 37°C. N52.E6 cells are usually passaged twice a week 1:4-5.
- The day before transfection, wash cells in PBS, detach cells in trypsin, and resuspend in culture medium. Count cells and plate 2 × 106 cells on a 6-cm dish. The cells should be ready for transfection at 60-80% confluency the following day.
Transfection
- Transfect N52.E6 cells in 6-cm dishes using 5µg of the SwaI-linearized pGS102#21 and the Superfect transfection kit. Perform transfection according to the manufacturer.
Comments
DNA transfections can also be performed using the calcium-phosphate coprecipitation method as described in a number of different manuals. The commercially available Effectene transfection Kit (Qiagen) also shows very high efficiency in transfection but should not be used for large-sized linearized plasmids.
Plaque Isolation and Preparation of High-Titer Helper Virus Stocks
- Six to 7 days posttransfection, harvest cells by scratching cells into medium, pellet cells for 10rain at 400 g, and resuspend in 2 ml Tris-buffered saline (TBS). Lyse cells by three cycles of freeze/thawing in dry ice/ethanol. Lysate can be stored at -80°C.
- Split 2 × 106 N52.E6 cells into 6-cm dishes the day before. Infect cells by removing medium, adding 3 ml fresh medium plus 0.2ml of the lysate. After 2h add 2ml medium. Five days after infection, harvest cells as described earlier and lyse by freeze/thawing.
- Split six 6-cm dishes N52.E6 the day before with 2 × 106 cells per dish. Dilute the lysate 1:100 in TBS and infect each two dishes with 1, 5, and 25 µl of the lysate.
- Three to 4h later, overlay cells with agarose: remove medium from infected cells and carefully add 10ml of prewarmed MEM-agarose solution. After the agarose solidifies, incubate at 37°C until plaques appear (usually 7-10 days).
- Pick well-isolated single plaques by punching out agar using a sterile Pasteur pipette and transfer agar to 0.2 ml TBS/10% glycerol. Plaques can be stored at -80°C.
- Infect 2 × 106 N52.E6 cells on 6-cm dishes with 100µl of the plaque lysate. After complete cytopathic effect (CPE) has occurred, harvest cells in 2 ml TBS and lyse by freeze/thawing.
- Amplify helper virus by first infecting 2 × 106 N52.E6 cells on a 6-cm dish with 1 ml of the lysate. Complete CPE should be visible 48 to 72h after infection. Harvest cells and titer helper virus in the lysate by infecting N52.E6 cells on a 6-cm dish (2 × 106 cells per dish, split the day before) with 30, 10, 5, and 1 µl (lysate 1:10 diluted).
- Infect ten 15-cm dishes N52.E6 cells (2 × 107 per dish) with the amount of lysate titered before and resulting in complete CPE after 48 h.
- Harvest cells 48 h after infection and centrifuge cells 10 min at 400 g. Dissolve pellet in 5 ml TBS. Lyse by freeze/thawing. Centrifuge for 15 min at 400g and 4°C.
Gradient Purification of Helper Virus
- Take supernatant and add TBS up to exactly 10ml. Add 5 g CsCl and mix gently. Be sure that CsCl is dissolved completely. Transfer lysate to SW41 centrifuge tube.
- Spin in Beckmann SW41 for 16-22h at 4°C and 32,000rpm.
- Collect the helper virus band by puncturing the tube with a needle and by drawing off the virus band in minimal volume.
- Add TBS/CsCl solution up to 10ml. Transfer to centrifuge tube and spin in a Beckmann SW41 as described earlier. Collect virus in minimal volume as before.
- Desalt virus using the PD-10 desalting column according to the manufacturer.
Titration of Helper Virus
- Determine particles and infectious units using the slot-blot protocol described later.
- Alternatively, determine the approximate amount of helper virus resulting in complete cpe of N52.E6 cells after 48 h.
DNA Preparation from CsCl-Purified Helper Virus Particles
In order to exclude helper virus rearrangement, viral DNA should be isolated from CsCl-purified particles. Take 200µl of gradient-purified and desalted vector. Isolate DNA using the QiaAmp DNA Minikit according to the manufacturer. Subsequent to ethanol precipitation, dissolve the pellet in 20µl AE buffer. Digest 10µl with the appropriate enzyme and run in an 0.8% agarose gel containing EtBr. As a positive control, double digest the corresponding helper virus plasmid with SwaI and the aforementioned enzyme.
Production of HC-Ad Vectors
Cloning of HC-Ad Vector Plasmids
General cloning reagents and equipment, as well as procedures, have been described in a number of cloning manuals. Thus, only important steps, observations, and suggestions are discussed in this section.
- The gene of interest including promoter and polyadenylation signal should be excisable in one piece.
- The fragment containing the gene of interest should be purified in an agarose gel using standard protocols (e.g., QIAEXII gel extraction kit, Qiagen Cat. No. 20021 for fragments <5 kb, electroelution for DNA fragments >5 kb). In order to improve the efficiency of cloning, it is important that the agarose gel does not contain EtBr. The DNA in the gel should never be exposed to a UV screen. Instead, the DNA fragments should be stained using Sybr-Gold (Molecular Probes Cat. No. S-11494) and visualized with a Dark Reader (e.g., Molecular Probes).
- For cloning, highly competent bacteria should be used (e.g., Stratagene XL2-Blue ultracompetent cells, Cat. No. 200150).
- Despite the size of 30-35 kb, the vector plasmid DNA can be isolated in a good amount and quality using standard plasmid DNA isolation protocols or kits.
- Plasmids should never be stored as glycerol stock but as DNA in TE either at 4°C or in ethanol at -20°C. Repeated freezing and thawing should be avoided. In addition, plasmid colonies on agar plates containing antibiotics should not be stored for more than 3 days.
Preparation of Plasmid DNA for Transfection
In order to release the left and right adenoviral termini (ITR), HC-Ad vector plasmids usually contain restriction sites flanking both ITRs. ITRs in the HC-Ad vector plasmids depicted in Fig. 2 are flanked by unique PmeI sites (or SnaBI site in pSTK142). The HCAd vector plasmid is digested and further purified as described earlier for the helper virus plasmid.
Culture of 73/29 Cells
Cre-expressing 73/29 cells are used for the production of HC-Ad vectors and are cultivated in αMEM containing 10% FBS and antibiotics, supplemented with 200µg/ml G418. 73/29 cells were usually passaged twice a week and diluted 1:4-5.
Amplification of HC-Ad Vectors
First Amplification/Transfection
- Seed 2 × 106 73/29 cells in one 6-cm dish the day before transfection.
- Transfect 5µg of PmeI-digested phenol/chloroform- purified and ethanol-precipitated HC-Ad vector plasmid using the Qiagen Superfect transfection kit according to the manufacturer. Incubate at 37°C in a CO2 incubator.
- Four to 16h after transfection, infect cells with 5 MOI of helper virus (multiplicity of infection).
- Forty-eight hours later, centrifuge infected cells (complete cpe should have occurred) for 10min at 400g. Dissolve pellet in 2ml TBS and lyse by freeze/thawing. Lysate can be stored at -80°C.
Second and Third Amplification
- Seed 2 × 106 73/29 cells in one 6-cm dish the day before.
- The next day infect cells with 1 ml of the lysate from the first amplification. At the same time, add 5 MOI of helper virus.
- Harvest the cells 48 h later and centrifuge for 10 min at 400g. Dissolve pellet in 2ml TBS and lyse by freeze/thawing.
- Infect 73/29 cells in one 6-cm dish (2 × 106 cells seeded the day before) with 1 ml of the lysate from the second amplification and coinfect with 5 MOI of helper virus.
- Again harvest cells 48 h after infection when complete cpe is visible and spin for 10 min at 400 g. Dissolve the pellet in 2 ml TBS and lyse three times by freeze/thawing.
Fourth and Fifth Amplification
- Seed 1-2 × 107 73/29 cells in one 15-cm dish the day before.
- Infect cells with half of the lysate from the third amplification and coinfect with 5 MOI of helper virus.
- Forty-eight hours later harvest cells and pellet for 10rain at 400g. Dissolve cells in 2 ml TBS and lyse three times by freeze/thawing.
- Infect two 15-cm dishes of 73/29 cells (2 × 107 cells per dish seeded the day before) with 2ml of the lysate from the fourth amplification and coinfect with 5 MOI of helper virus.
- Fourty-eight hours later harvest cells and pellet for 10rain at 400g. Dissolve cells in 2 ml TBS and lyse three times by freeze/thawing. Centrifuge for 15min at 400g and 4°C. Keep the supernatant for further amplifications. Keep the pellet for further testing (see later).
Titering Amplifications
After the fifth amplification it is important to titer the amount of vector present in the lysate. In case HCAd vectors express a reporter gene (e.g., β-gal, EGFP), the vector can be titered easily in a reporter gene assay. Figure 3 shows blue forming unit (bfu) titers in amplifications of AdGS46 (see Fig. 1). However, most HCAd vectors do not contain a reporter gene and thus the ratio of helper and vector genomes and possible vector genome rearrangements should be tested using DNA isolated from infected cells.
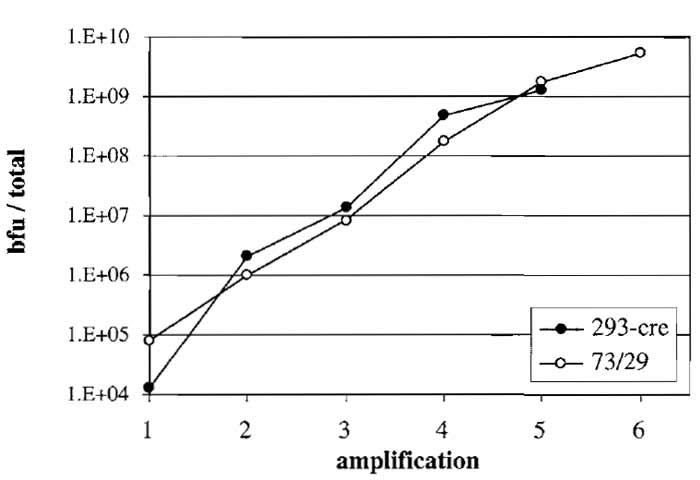 |
| FIGURE 3 Amplification of HC-Ad vector AdGS46 using helper
virus AdGS102#21 and a HEK293-based (293-cre) or N52.E6-based (73/29) Cre-expressing producer cell line. Both cell lines were transfected with PmeI-digested pGS46 and, at the same time, infected with helper virus (amplification 1). AdGS46 was serially amplified in both cell lines. Titers in each amplification were assayed by infecting N52.E6 cells with aliquots of the lysate followed by staining for β-galactosidase and quantitation as blue forming units (bfu). |
- Resuspend the pellet obtained in step 14 in 500µl TBS.
- Take 100µl, add 50µl TE, 20µl 10% SDS, 20µl proteinase K, 10µl 0.5M EDTA, vortex, and incubate for 2h at 37°C.
- Extract DNA by phenol/chloroform extraction and subsequent precipitation with ethanol.
- Digest 1 µg DNA with an appropriate enzyme and add RNase to the digest. As a useful positive control, double digest 200ng of the corresponding HC-Ad vector plasmid with PmeI and the aforementioned enzyme. Separate fragments in an 0.8% agarose gel containing EtBr (run gel at low voltage, preferably overnight).
Comments
At this stage of amplification the majority of DNA within the producer cell is vector DNA with only little helper virus contamination. Therefore, if the amplification was successful, only bands corresponding to the vector should be visible with a weak background of the helper virus bands and a faint smear of cellular DNA. In addition, rearranged vector genomes can be detected. If there are no or only weak vector bands visible, one or two additional amplifications can be performed. However, it is not recommended to carry out more than seven amplifications, as the risk of outgrowth of mutated helper virus and vector rearrangements increases significantly.
Preparation of Gradient-Purified HC-Ad Vector
- Plate ten 15-cm dishes of 73/29 cells with 2 × 107 cells per dish.
- The next day infect cells with the lysate from the last amplification (supernatant obtained in step 14) and 5 MOI of helper virus. Incubate for 48 h until complete cpe has occurred.
- Harvest the cells by spinning for 15min at 400g. Resuspend cells in 5ml TBS and lyse by freeze/thawing. Spin for 15 min at 400 g.
- Split supernatant into two sterile 15-ml tubes, fill up to 10 ml with TBS, and add 5 g CsCl to each tube. Mix carefully until CsCl is dissolved. Transfer to two ultraclear centrifuge tubes.
- Spin in a Beckmann SW41 for 16-22h at 4°C and 32,000rpm.
- In optimal vector preparations, only one band containing vector particles should be visible. If helper virus contamination is high, a second helper virus particle band is visible in addition to the upper vector particle band separated by only a few millimeters. The upper vector band can be collected by puncturing the tube with a needle just below the vector band and drawing off the vector band in a small volume. Combine vector particles from both tubes. It is also possible to start vector purification by a CsCl step gradient followed by one or two continuous gradients.
- Add TBS/CsCl solution up to 10 ml. Transfer to a new ultraclear centrifuge tube and spin in a Beckmann SW41 as described previously. Collect the vector in a small volume as described earlier.
- Desalt vector particles using the PD-10 desalting column according to the manufacturer.
DNA Preparation from CsCl-Purified Vector Particles
In order to finally exclude vector rearrangement vector DNA should be isolated from CsCl-purified particles as described for extraction of helper virus DNA. The DNA should be digested with the appropriate enzyme and run on a 0.8% agarose gel containing EtBr. As a positive control, double digest the corresponding HC-Ad vector plasmid with PmeI and the aforementioned enzyme.
Troubleshooting
| Problem | Comments and suggestions |
| Low helper virus titer | Helper virus might be impaired in DNA replication. Test for rearrangements in the helper virus genome. Alternatively, use an E3-containing helper virus |
| Instability of HC-Ad vector cloning plasmids |
pBluescript containing large inserts tend to rearrange. DNA should be isolated from colonies that have been transformed the day before. Increasing the ampicillin concentration to 100µg/ml is recommended |
| Poor transfection efficiency | Transfection efficiency should be tested and optimized using a linearized HC-Ad vector plasmid expressing a reporter gene (β-gal, EGFP). Transfection can be inhibited by traces of phenol present in the linearized DNA. Purify DNA by an additional ethanol precipitation |
| Adding 5 MOI of helper virus during amplification does not results in complete cpe after 48h |
Because the amount of vector increases during amplification, it might also be necessary to increase the amount of helper virus. Because amplification efficiency depends on the producer cell and the type of helper virus used, it is highly recommended to standardize amplification using a reporter gene expressing vector for each type of producer cell and helper virus |
| Vector rearranges during amplification |
Make sure that the vector size ranges between 27 and 35 kb. Smaller vectors tend to rearrange |
| No vector obtained even after sever or eight rounds of amplification |
Make sure your that the transgene is not toxic for producer cells and that the vector did not rearrange during amplifications. Another possible explanation for a failed vector amplification is helper virus outgrowth due to deletion of a loxP site |
| High helper virus content in CsCl- purified vector |
There are several possible explanations for high helper virus contamination: (1) The producer cell expresses low or no Cre-recombinase. Grow cells for several passages in the selection medium (e.g., G418). (2) One loxP site in the helper virus is lost due to recombination with vector genomes. Test for the presence of both loxP sites using PCR and primers corresponding to sequences flanking both loxP sites. (3) The helper virus turned into RCA in case HEK293-based producer cells have been used. Test for presence of RCA using PCR |
| Helper and vector particle bands do not separate clearly |
Especially if vector yield is low, the vector or helper virus band is hardly visible. Using a fiber-optic gooseneck lamp when drawing off the band from the CsCl gradient simplifies this procedure significantly |
III. COMPLETE CHARACTERIZATION OF HC-Ad VECTOR PREPARATIONS
Materials
First Day: Seeding Cells
Cell lines: A549 (ATCC number: CCL-185) or, alternatively, HeLa cells (ATCC number: CCL-2)
Supplies: 24-well tissue culture plates
Second Day: Transduction of Cells with HC-Ad Vectors
Reagents and solutions: Tris-buffered saline (TBS, see Section II.)
Third Day: Slot Blotting of Cell and Vector Lysates; Hybridization
Equipment
Shaker (e.g., Scientific Industries : Vortex Genie Model G-560E)
Vacuum pump (e.g., Biometra: Typ PM12640-026.3)
Slot-blot apparatus (e.g., Amersham Biosciences: Hoefer PR648)
Hybridisation oven (e.g., Biometra: Duo-Thermo- Oven OV5)
Supplies
Positively charged nylon membrane (Pall: Biodyne B, 0.45 µm)
50µCi [32P] dCTP
RediPrime II DNA labeling kit (Amersham- Biosciences)
Reagents and Solutions
Phosphate-buffered saline (PBS): 6.46mM Na2HPO4, 1.47mM KH2PO4, 137mM NaCl, 2.7mM KCl, pH 7.4, autoclave
PBS/20mM EDTA: Add EDTA from aqueous stock solution (0.5M, pH 8, see Section II) to PBS
0.8N NaOH: Dissolve 32g NaOH pellets in 1 liter deionized H2O, always prepare fresh
(Pre-)hybridisation solution: 2 × SSC (300mM NaCl, 30mM Na-citrate, pH 7.0) containing 10% dextran sulfate, 1% SDS, 0.5% milk powder (low fat), and 0.5 mg/ml salmon sperm DNA
DNA Templates for Generation of Probes
Template for generation of HC-Ad vector-specific probe: This should be a purified PCR fragment of 600-1200bp length. Usually the PCR fragment is derived from the stuffer DNA or the transgene expression cassette of the HC-Ad vector plasmid. This probe is used for determination of both the number of infectious units and total particles. Alternatively, an Ad5-ITR probe can be used that is generated by PCR with the primers P1 (5'-CATCATCAATAATATACCTTATTTTG- 3') and P2 (5'- AACGCCAACTTTGACCCGGAACGCGG-3') from a plasmid containing at least the left Ad5 ITR.
Template for generation of helper virus-specific probe: This is a PCR fragment comprising Ad5 nucleotides 31042-32390 (Ad5 fiber gene) obtained from a plasmid containing the fiber gene of Ad5 with the primers P3 (5'-ATGAAGCGCGCAAGACCGTCTG- 3') and P4 (5'-CCAGATATTGGAGCCAAACTGCC- 3').
Plasmids for Generating Standard Curves
Comment: It is recommended to prepare a large stock of standard plasmids at appropriate concentrations (1-3E+06 copies/µl) and keep it frozen (-20°C).
The plasmid used to generate the standard curve for determining infectious and total particle contents of HC-Ad vector preparations is usually the HC-Ad vector shuttle plasmid that was used to generate the vector and/or the PCR template for the probe. The concentration of this standard plasmid should be determined as accurately as possible.
The plasmid used to generate the standard curve for determining the helper virus content of HC-Ad vector preparations is usually a shuttle plasmid for the generation of El-deleted vectors and should contain the Ad5 fiber gene. The concentration of this standard plasmid should be determined as accurately as possible.
Fourth Day: Exposition of PhosphorScreen and Signal Quantification
Equipment
PhosphorScreen (e.g., Amersham-Biosciences/Molecular Dynamics: Kodak Storage Phosphor Screen SO230)
PhosphorImager (e.g., Amersham-Biosciences/Molecular Dynamics: Storm 860)
Quantification Software (e.g., Amersham- Biosciences/Molecular Dynamics: ImageQuaNT)
Supplies
Saran wrap
Reagents and Solutions
Wash buffer I: 2 × SSC (300 mM NaCl, 30 mM Na-citrate, pH 7.0) containing 0.1% SDS
Wash buffer II: 0.1 × SSC (15mM NaCl, 1.5mM Nacitrate, pH 7.0) containing 0.1% SDS
Procedure
First Day: Seeding Cells
- Calculate the amount of wells needed: [No. of vectors to be titered × 6] +10.
- Seed 1-2E+05 A549 or HeLa cells per well.
- Let cells attach overnight in 1 ml of medium (37°C).
Second Day: Transduction of Cells with HC-Ad Vectors
- Dilute the vectors 1:20 with TBS.
- Aspirate medium from the cells and replace with 300 µl fresh medium.
- Transduce the cells with 2, 10, and 20 µl of the vector dilutions in duplicates (six wells per vector).
- Leave 10 wells untransduced to generate a standard curve for infectious particles.
- Incubate cells for 12-14h at 37°C.
Third Day: Slot Blotting of Cell and Particle Lysates; Hybridization
Preparation of Cell Lysates from Transduced Cells for Determining the Number of Infectious Particles
- Aspirate the medium from transduced cells.
- Wash the cells once with 1 ml prewarmed PBS (37°C) per well.
- Incubate the cells with 1 ml prewarmed PBS for 5 min at 37°C.
- Aspirate PBS and add 200µl of PBS/EDTA per well. Incubate for 10rain at 37°C.
- Detach the cells by pipetting up and down several times and transfer them into 1.5-ml reaction tubes.
- Add 200µl 0.8N NaOH per reaction tube, mix thoroughly, and incubate at room temperature for 30rain. During incubation mix thoroughly every 10 min.
Preparation of Cell Lysates for Generation of a Standard Curve for Infectious Particles
Comment: The debris in the cell lysate that is slot blotted onto the membrane will influence the signal intensity due to partial blocking of the membrane. Therefore, to obtain reliable standard curves for determining the number of infectious HC-Ad vector particles, it is absolutely required to mix the standard plasmid with untransduced cells and prepare lysates in the same way as for transduced cells before blotting.
- Aspirate the medium from untransduced cells.
- Wash the cells once with 1 ml prewarmed PBS (37°C) per well.
- Incubate the cells with 1 ml prewarmed PBS for 5 min at 37°C.
- Aspirate PBS and add 200µl of PBS/EDTA per well. Incubate for 10min at 37°C.
- Detach the cells by pipetting up and down several times and transfer them into 1.5-ml reaction tubes.
- Add 1E+06, 5E+06, 1E+07, 5E+07, and 1E+08 copies of the standard plasmid in duplicates to the cells in the reaction tubes. Do not to add more than 40 µl.
- Add 200µl 0.8N NaOH per reaction tube, mix thoroughly, and incubate at room temperature for 30min. During incubation mix thoroughly every 10 min.
Preparation of Particle Lysates for Determining the Number of Total Particles
Comment: Particle lysates for determining the number of total and helper virus particles do not contain any debris that blocks the membrane. Therefore, standard plasmids are prepared in PBS/EDTA and treated the same way as particle lysates.
- Dilute the vectors 1:600 with TBS.
- Transfer in duplicates 2, 10, and 20µl of the vector dilutions into 1.5-ml reaction tubes and fill up to 200µl with PBS/EDTA.
- Add 200µl 0.8N NaOH per reaction tube, mix thoroughly, and incubate at room temperature for 30min. During incubation mix thoroughly every 10 min.
Preparation of a Standard for Determining the Number of Total Particles
- Prepare 1E+06, 5E+06, 1E+07, 5E+07, and 1E+08 copies of the standard plasmid in 200µl PBS/EDTA in duplicates in 1.5-ml reaction tubes.
- Add 200µl 0.8N NaOH per reaction tube, mix thoroughly, and incubate at room temperature for 30min. During incubation mix thoroughly every 10 min.
Preparation of a Standard for Determining the Number of Helper Virus Particles
- Prepare 1E+06, 5E+06, 1E+07, 5E+07, and 1E+08 copies of the standard plasmid in 200µl PBS/EDTA in duplicates in 1.5-ml reaction tubes.
- Add 200µl 0.8N NaOH per reaction tube, mix thoroughly, and incubate at room temperature for 30min. During incubation mix thoroughly every 10 min.
Slot Blotting of Lysates and Standard onto Positively Charged Nylon Membranes
Comment: The cell lysates and particle lysates for determining the number of infectious and total HCAd- vector particles can be blotted onto the same membrane, as the same probe is used for hybridization. Particle lysates for determining the number of helper virus particles must be blotted onto a separate membrane and will be hybridized with a different probe.
- Soak an appropriately sized membrane 2min in 0.4 N NaOH.
- Assemble the slotbot apparatus with the membrane in correct position.
- Connect the slot blot apparatus to the vacuum pump and apply vacuum (max -400mbar).
- Check tightness by applying 200 µl deionized water to one slot.
- Release vacuum.
- Pipet two-thirds of each lysate into separate slots.
- Apply vaccum starting with-400 mbar and slowly inrease up to -800mbar until lysates pass the membrane completely.
- Release vacuum and disassemble slot blot apparatus.
- Rinse the membrane twice in 2 × SSC (5 min).
- Bake the membrane at 80°C for 20min.
- Boil prehybridization buffer for 10-15min in a water bath. Cool down to room temperature on ice. Use 20ml of prehybridization buffer per membrane.
- Prehybridize membrane at 68°C in 20ml prehybridization buffer (1-2 h).
- Label 100 ng of the DNA probe with [32P]dCTP and add it to the prehybridization buffer.
- Hybridize for 12-15h.
Fourth Day: Exposition of PhosphorScreen and Signal Quantification
- Prewarm wash buffers I and II to 68°C in a water bath.
- Wash membrane twice with prewarmed wash buffer I for 10min at 68°C.
- Wash membrane with prewarmed wash buffer II for 10 min at 68°C.
- Wash membrane with wash buffer II for 5min at room temperature.
- Air dry membrane.
- Start exposition of the PhosphorScreen (usually 2-3h).
- Read the phosphor screen with PhosphorImager and quantify signals.
- Calculate standard curve from signal intensities and use linear regression to quantify the signal intensities of the vector samples. The coefficient of correlation for the standard should be above 0.99.
- Calculate the inverse bioactivity [No. of total particles/ No, of infectious particles].
IV. RESULTS
Figure 4a shows a typical result for determination of the number of infectious particles of different HCAd vector preparations. Figure 4b shows the standard curve obtained from the signal intensities of the standard plasmid. The parameters of the standard curve (slope m and intercept b) were used to calculate the number of infectious particles of the vector preparations HC-AdV1 to HC-AdV4. These titers are given in Table I. Slot blots to determine the number of total particles and helper virus particles look similar and are omitted for brevity. The inverse bioactivity (number of total particles divided by number of infectious particles) usually is between 10 and 200, and the helper virus contamination is between 0.1 and 2%.
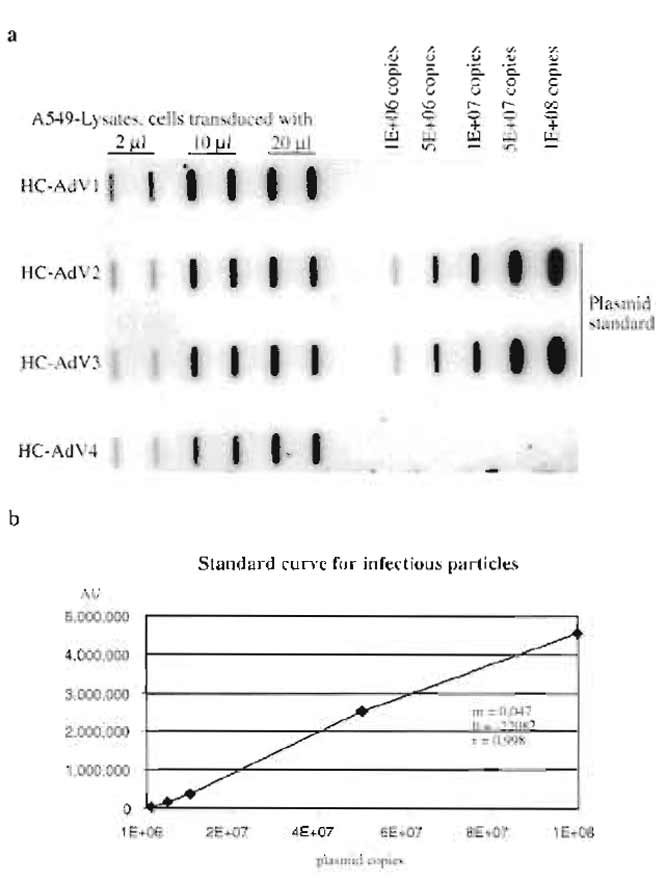 |
| FIGURE 4 (a) Slot blot to determine the number of infectious
units of different HC-Ad-vector preparations (HC-AdV1 to HCAdV4). (b) The standard curve obtained from signal intensities of the standard plasmid, r, coefficient of correlation; b, intercept, and m, slope. These parameters were used to calculate the number of infectious particles for HC-AdV1 to HC-AdV4. |
 |
References
Fallaux, F. j., Bout, A., van der Velde, I., van den Wollenberg, D. J., Hehir, K. M., Keegan, J., Auger, C., Cramer, S. J., van Ormondt, H., van der Eb, A. J., Valerio, D., and Hoeben, R. C. (1998). New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum. Gene Ther. 9, 1909-1917.
Gao, G. P., Engdahl, R. K., and Wilson, J. M. (2000). A cell line for high-yield production of El-deleted adenovirus vectors without the emergence of replication-competent virus. Hum. Gene Ther. 11, 213-219.
Graham, E L., Smiley, J., Russel, W. C., and Nairn, R. (1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36, 59-74.
Hartigan-O'Connor, D., Barjot, C., Crawford, R., and Chamberlain, J. S. (2002). Efficient rescue of gutted adenovirus genomes allows rapid production of concentrated stocks without negative selection. Hum. Gene Ther. 13, 519-531.
Hilgenberg, M., Schnieders, F., L6ser, P., and Strauss, M. (2001). System for efficient helper-dependent minimal adenovirus construction and rescue. Hum. Gene. Ther. 12, 643-657.
Kreppel, E, Biermann, V., Kochanek, S., and Schiedner, G. A DNAbased method to assay total and infectious particle contents and helper virus contamination in high-capacity adenoviral vector preparations. Hum. Gene Ther. 13, 1151-1156.
Ng, P., Beauchamp, C., Evelegh, C., Parks, R., and Graham, E L. (2001). Development of a FLP/frt system for generating helperdependent adenoviral vectors. Mol. Ther. 3, 809-815.
Palmer, D., and Ng, P. (2003). Improved system for helper-dependent adenoviral vector production. Mol. Ther. 11, 504-511.
Parks, R. J., Bramson, J. L., Wan, Y., Addison, C. L., and Graham, E L. (1999). Effects of stuffer DNA on transgene expression from helper-dependent adenovirus vectors. J. Virol. 73, 8027- 8034.
Parks, R. J., Chen, L., Anton, M., Sankar, U., Rudnicki, M. A., and Graham, F. L. (1996). A helper-dependent adenovirus vector system: Removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. USA 93, 13,565-13,570.
Parks, R. J., and Graham, E L. (1997). A helper-dependent system for adenovirus vector production helps define a lower limit for efficient DNA packaging. J. Virol. 7, 3293-3298.
Sakhuja, K., Reddy, P. S., Ganesh, S., Cantaniag, E, Pattison, S., Limbach, P., Kayda, D. B., Kadan, M. J., Kaleko, M., and Connelly S. (2003). Optimization of the generation and propagation of gutless adenoviral vectors. Hum. Gene Ther. 14, 243-254.
Sandig., V., Youil, R., Bett, A. J., Franlin, L. L., Oshima, M., Maione, D., Wang, E, Metzker, M. L., Savino, R., and Caskey, C. T. (2000). Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc. Natl. Acad. Sci. USA 97, 1002-1007.
Schiedner, G., Hertel, S., and Kochanek, S. (2000). Efficient transformation of primary human amniocytes by E1 functions if Ad5: Generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 11, 2105-2116.
Schmid, S. I., and Hearing, P. (1998). Cellular components interact with adenovirus type 5 minimal DNA packaging domains. J. Virol. 72, 6339-6347.
Thomas, C. E., Schiedner, G., Kochanek, S., Castro, M. G., and L6wenstein., P. R. (2000). Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with highcapacity, adenovirus vectors: Toward realistic long-term neurological gene therapy for chronic diseases. Proc. Natl. Acad. Sci. USA 97, 7482-7487.
Umana, P., Gerdes, C. A., Stone, D., Davis, J. R., Ward, D., Castro, M. G., and L6wenstein, P. R. (2001). Efficient FLPe recombinase enables scalable production of helper-dependent adenoviral vectors with negligible helper-virus contamination. Nature Biotechnol. 19, 582-585.
Support our developers




