Cell Proliferation Assays:Improved Homogeneous Methods Used to Measure the Number of Cells in Culture
Over the last decade several improvements have been made in assay technology to enable miniaturization and more efficient measurement of the number of cells present in microwell plates. A variety of different methods have been optimized for convenient use in multiwell formats, making it easier to do large numbers of assays. The most significant improvement in efficiency has been the development of homogeneous "add, mix, and measure" assay formats compatible with robotic automation for high-throughput screening (HTS) of test compounds.
Making a choice among available assay formats often depends on the preference for which marker is measured or the level of sensitivity required. Homogeneous assays are now available to measure total cell number, viable cell number, the number of dead cells present in a population, or the number of cells undergoing apoptosis. For many experimental systems the most useful information is the number of viable cells at the end of a treatment period. The parameter used most conveniently to determine the number of viable cells in culture is measurement of an indicator of active metabolism.
II. MATERIALS
RPMI 1640 culture medium containing 15 mM HEPES (Cat. No. R-8005), 2-mercaptoethanol (Cat. No. M-7154), trypan blue solution (0.4% Cat. No. T-8154), and phenazine ethosulfate (PES; Cat. No. P-4544) are from Sigma. Fetal bovine serum (Cat. No. SH30070) is from Hyclone. Ninety-six-well plates with opaque white walls and clear bottoms (Cat. No. 3610) are from Corning.
III. INSTRUMENTATION
The Model MTS-4 plate shaker obtained from IKA Works, Inc. is used to mix the contents of the 96-well plates. Absorbance is recorded using a Molecular Devices Vmax plate reader spectrophotometer. Luminescence is recorded using a Dynex MLX plate-reading luminometer. Fluorescence is recorded using a Labsystems Fluoroscan Ascent fluorescence plate reader fitted with a 560 (excitation) and 590 (emission) filter pair.
IV. PROCEDURES
The assay plates for all four procedures are prepared in an identical manner using stock cultures of the IL-6-dependent B9 hybridoma cell line. B9 cells are cultured in RPMI 1640 containing 15 mM HEPES and 50µM 2-mercaptoethanol supplemented with 10% fetal bovine serum (assay medium) and 2 ng/ml of IL- 6 (specific activity 168 U/ng protein, assigned by direct comparison to the interim reference standard #88/514 from the National Institute of Biologic Standards and Controls). Cell cultures are maintained at densities between 2 × 104 and 3 × 105/ml in a humidified chamber at 37~ with 5% CO2.
Serial two-fold dilutions of IL-6 are prepared in assay medium without IL-6 so 20-µl/well additions would contain a final concentration range of 0-2.0ng/ml. An equivalent volume of assay medium without IL-6 is added to sets of negative control wells. Assay plates are cultured for 72h at 37°C with 5% CO2 before processing with each of the cell number assays.
A. MTS Tetrazolium Reduction Assay
Viable cells reduce tetrazolium compounds into intensely colored formazan products that can be detected as an absorbance change with a spectrophotometer. The amount of formazan color produced is directly proportion to the number of viable cells in standard culture conditions. Cells rapidly lose the ability to reduce tetrazolium compounds shortly after death, which enables tetrazolium reduction to be used as an indicator of viable cell number. The first MTT tetrazolium reduction cell proliferation assay was described two decades ago (Mosmann, 1983). Upon cellular reduction, the MTT tetrazolium reagent results in the formation of a formazan precipitate and requires the addition of a solubilizing agent to generate a solution suitable for recording absorbance.
The combination of MTS + PES provides an assay that requires only a single reagent addition to the cell culture wells and results in a homogeneous "add, incubate, and measure" assay format. An additional advantage of using aqueous soluble tetrazolium assays is that data can be recorded from the same plate at various intervals after the addition of MTS + PES, which simplifies the optimization of the incubation period during characterization of the effects of particular compounds on cells (Fig. 1). The sensitivity of the MTS assay is dependent on cell type, but it is usually adequate for detecting the number of cells used commonly in microwell plates. Typically, the MTS assay can detect fewer than 1000 viable cells/well in the 96- well plate format or fewer than 250 cells/well in 384- well plates. For additional background information, refer to Promega Technical Bulletin #245.
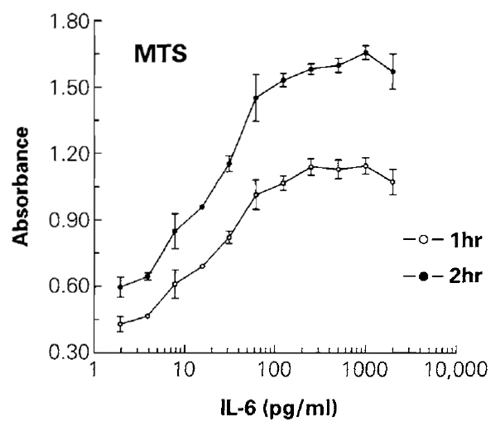 |
| FIGURE 1 The effect of different concentrations of IL-6 on cell number was determined by measuring MTS reduction to the colored formazan product. Absorbance at 490nm was recorded after 1 h of incubation with the MTS + PES reagent. Plates were returned to the cell culture incubator for an additional hour before recording the 490-nm absorbance after a total of 2h of incubation. The 490-nm absorbance from control wells of cells without IL-6 (not shown on the log scale) was 0.39 for 1 h of incubation and 0.54 for 2 h of incubation. Values represent the mean ± standard deviation from four replicate wells. |
Steps
- Warm the CellTiter 96 AQueous one solution reagent to 37°C (Note: The reagent contains 2mg/ml MTS tetrazolium and 300µM PES electron transfer reagent in a solution of phosphate-buffered saline. This solution is photolabile and should be stored protected from light.)
- Remove the multiwell assay plate from the cell culture incubator and transfer to a laminar flow hood.
- Add 20µl of CellTiter 96 AQueous one solution reagent to the culture medium in each well using a multichannel pipette and aseptic conditions.
- Mix the contents of the culture wells using a plate shaker to ensure uniform suspension of MTS reagent in the culture medium.
- Return assay plate to a 37°C cell culture incubator for a period of 1 to 4 h, depending on the number and metabolic activity of cells being used.
- Transfer assay plate to a multiwell plate-reading spectrophotometer, shake plate for 10s (using the instrument's on-board plate shaking function) to ensure a uniform solution in the assay wells, and record absorbance at 490nm.
- Plot 490nm absorbance vs cell number.
B. ATP Assay
The measurement of ATP has become widely accepted as a valid indicator of the number of viable cells present in culture (Ekwall et al., 2000). Under cell culture experimental conditions that do not alter metabolism drastically, the amount of ATP is directly proportional to the number of viable cells (Crouch et al., 1993). Historically, sample preparation for ATP assays has been a multistep process requiring inactivation of endogenous ATPases (known to interfere with measurement of ATP) and neutralization of the acidic extract prior to addition to a luciferasecontaining reaction mixture (Lundin et al., 1986; Stanley, 1986). Firefly luciferase purified from Photinus pyralis has been used most often as a reagent for ATP assays (Lundin et al., 1986; Crouch et al., 1993). Unfortunately, the native form of luciferase has only moderate stability in vitro and is sensitive to its chemical environment, e.g., pH and detergents, thus limiting its usefulness for developing a robust homogeneous ATP assay.
Preparation of CellTiter-Glo Reagent
- Allow a vial of the lyophilized substrate and a bottle of frozen assay buffer to equilibrate to ambient temperature (22°C).
- Add 10ml of assay buffer to the substrate to reconstitute the lyophilized cake and form the CellTiter-Glo reagent and mix gently to dissolve. The reagent is a buffered solution containing detergents to lyse cells, ATPase inhibitors to stabilize the ATP released from the lysed cells, luciferin as a substrate, and luciferase to generate a bioluminescent signal proportional to the amount of ATP present in the cell lysate.
- Remove the multiwell assay plate from the cell culture incubator and equilibrate to ambient temperature for approximately 20-30min. (Note: Transferring eukaryotic cells from 37°C to ambient temperature for the length of time required for temperature equilibration has little affect on cell viability or ATP content.)
- Add a volume of CellTiter-Glo reagent equal to the volume of cell culture medium present in each well using a multichannel pipette. (Note: For 384-well plates containing 25 µl of culture medium, add 25 µl of reagent.)
- Mix the contents of the assay wells to ensure uniform distribution of the reagent in the culture medium and to speed cell lysis.
- Allow the assay plate to stand at ambient temperature for 10 min.
- Record luminescence using a multiwell platereading luminometer.
- Plot luminescence vs cell number (Fig. 2).
Lactate dehydrogenase is a cytoplasmic enzyme that has been used as a marker of cell damage in vitro because the enzymatic activity is relatively stable in cell culture medium and can be measured easily after leakage out of cells with a compromised membrane (i.e., nonviable cells). The LDH assay is performed most commonly as a cytotoxicity assay by measuring the enzymatic activity from a sample of culture medium removed from the treated population of cells. Most assay methods require transfer of an aliquot of culture medium (without cells) into a separate assay vessel because the reagent formulation would damage living cells, resulting in the release of additional LDH (Korzeniewski and Callewaert, 1983). Reactions often proceed for 30min and result in a colorimetric signal (Decker and Lohmann-Matthes, 1988).
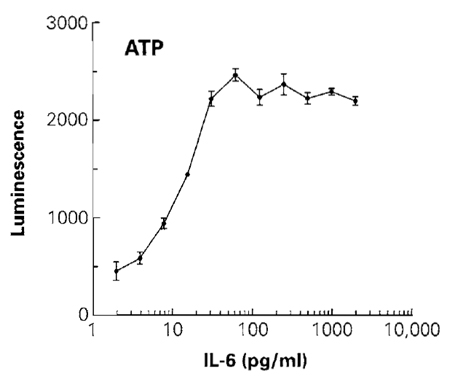 |
| FIGURE 2 The effect of different concentrations of IL-6 on cell number was determined by measuring the total amount of ATP. Luminescence was recorded 10min after addition of the CellTiter-Glo reagent. The luminescence of control wells of cells without IL-6 (not shown on the log scale) was 260 relative light units. Values represent the mean ± standard deviation from four replicate wells. |
Recent improvements in LDH assay performance have been accomplished by using more sensitive fluorescent reporter molecules and by formulating the assay reagents in a physiologically balanced buffer that is not harmful to viable cells. These improvements enabled the development of a rapid homogeneous cytotoxicity assay format to detect the number of damaged cells directly in cell culture wells containing a mixed population of viable and nonviable cells. The reagent used to perform the coupled enzymatic assay is a buffered solution containing lactate as a substrate and NAD+ as a cofactor to drive the LDH reaction. The reagent also contains the enzyme diaphorase to catalyze the NADH-driven reduction of resazurin into the fluorescent resorufin product.
As illustrated in the following example procedure, the total number of cells in culture (i.e., viable and nonviable) also can be estimated using an LDH assay by measuring total enzymatic activity from the entire population of cells. The cytotoxicity assay format is modified to detect total LDH in cultures by including a cell lysis step in the procedure utilizing a detergent that is compatible with the LDH assay chemistry. The detection sensitivity and linear range of the fluorescent LDH assay are typically 800-50,000 cells/well in the 96-well plate format, with sensitivity improving to 200 cell/well in 384-well plates. For additional background information, refer to Promega Technical Bulletin #306.
- Allow a vial of the lyophilized substrate and a bottle of frozen assay buffer to equilibrate to ambient temperature (22°C).
- Add 11ml of assay buffer to the substrate to reconstitute the lyophilized cake and form the CytoTox-ONE reagent and mix gently to dissolve. Protect the reagent from direct light.
Steps
- Remove the multiwell assay plate from the cell culture incubator.
- Add 2µl of the lysis solution component of the kit (i.e., 9% Triton X-100) to each 100µl of culture medium to lyse the cells and release LDH into the surrounding medium. (Note: If it is inconvenient to pipette very small volumes, a 1:5 dilution of lysis solution may be prepared using water so 10µl can be dispensed into each well.)
- Shake the plate for approximately 10s to ensure uniform distribution of the contents of the wells and to ensure complete cell lysis.
- Equilibrate the plate to ambient temperature (approximately 20-30 min).
- Add a volume of CytoTox-ONE reagent equal to the volume of cell culture medium present in each well using a multichannel pipette. (Note: For 384-well plates containing 25µl of culture medium, add 25 gl of reagent to each well.)
- Mix the contents of the assay wells for 30s to ensure uniform distribution of the reagent in the culture medium.
- Allow the assay plate to incubate at ambient temperature for 10-15min for the LDH reaction to proceed.
- Add 50µl to each well of the stop solution [3% (w/v) sodium dodecyl sulfate] provided as a component of the CytoTox-ONE assay kit. (Note: Add 12.5µl stop solution for the 384-well plate format containing 25µl of cells in culture medium and 25µl of CytoTox- ONE reagent.)
- Record the fluorescence of resorufin using a multiwell plate reading fluorometer fitted with a filter set for 560-nm excitation and 590-nm emission wavelengths. (Note: The spectra of resorufin will enable a variety of filter sets to be used. Data can be collected using excitation filters in the range of 530-570nm and emission filters in the range of 580-620nm.)
- Plot fluorescence vs cell number (Fig. 3).
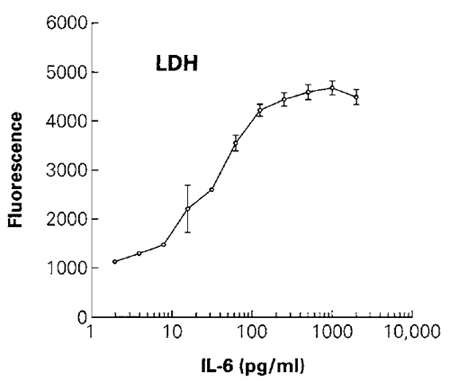 |
| FIGURE 3 The effect of different concentrations of IL-6 on cell number was determined by measuring the total amount of LDH from the lysed population of cells. Cells were lysed by the addition of 2µl/well of 10% (v/v) Triton X-100 in phosphate-buffered saline prior to addition of the CytoTox-ONE reagent. The reaction was stopped by addition of the stop solution, and fluorescence (560 excitation/590 emission) was recorded. The fluorescence value of control wells of cells without IL-6 (not shown on the log scale) was 1081 relative fluorescence units. Values represent the mean ± standard deviation from four replicate wells. |
D. Resazurin Reduction Assay
Resazurin is a redox dye that can be reduced by cultured cells to form resorufin. Resazurin is dark blue and has little intrinsic fluorescence until it is reduced to the pink resorufin product. The spectral properties of resorufin allow the molecule to be detected using either fluorescence or absorbance; however, fluorescence is the preferred method because it provides greater sensitivity.
The resazurin reduction assay is based on the ability of metabolically active living cells to convert a redox dye (resazurin) into a fluorescent end product (resorufin). Resazurin can enter living cells where it becomes reduced, and the resazurin product, which is also permeable, can be found in the cell culture medium (O'Brien et al., 2000). The specific cellular mechanisms responsible for the reduction of resazurin are unknown (Gonzales and Tarloff, 2001), but probably involve reactions generating reducing equivalents such as NADH. Nonviable cells lose metabolic capacity rapidly and do not reduce resazurin to generate a fluorescent signal. The resazurin substrate is soluble in phosphate-buffered saline compatible with preparation of a reagent for direct addition to cell cultures. The homogeneous assay procedure involves addition of a single resazurin-containing reagent directly to cells cultured in serum-supplemented medium. After an incubation step, data are recorded using either a platereading fluorometer (preferred method) or a spectrophotometer. The reagent is generally nontoxic to cells, allowing extended incubation periods in some situations. Fluorescence data can be recorded at various intervals after the addition of resazurin, which simplifies optimization of the incubation period during characterization of the effects of particular compounds on cells (Fig. 4). Longer incubation periods may result in increased detection sensitivity; however, there may be a loss in the linear range of response. Assay sensitivity and range depend on cell type and metabolic capacity, but are typically between 200 and 50,000 cells/well in 96-well plates. For additional background information, refer to Promega Technical Bulletin #317.
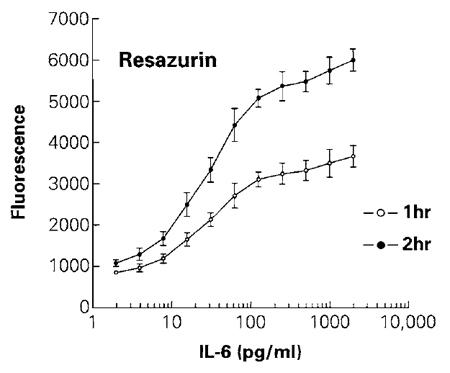 |
| FIGURE 4 The effect of different concentrations of IL-6 on cell number was determined by measuring the ability of viable cells to reduce resazurin into the fluorescent resorufin. Fluorescence was recorded after 1 h of incubation with the CellTiter-Blue reagent. Plates were returned to the cell culture incubator for an additional hour before recording fluorescence a second time after a total of 2h of incubation. The average fluorescence from control wells of cells without IL-6 (not shown on the log scale) was 814 for 1 h of incubation and 1017 for 2 h of incubation. Values represent the mean ± standard deviation from four replicate wells. |
Steps
- Thaw CellTiter-Blue reagent containing resazurin and warm to 37°C.
- Remove the multiwell assay plate from the cell culture incubator and transfer to a laminar flow hood.
- Add 20µl CellTiter-Blue reagent to each well containing 100µl culture medium using a multichannel pipette and aseptic conditions. (Note: For 384- well plates containing 25µl of culture medium, add 5µl/well of CellTiter-Blue reagent.)
- Mix the contents of the assay wells for 10s to ensure uniform distribution of the reagent in the culture medium.
- Return assay plate to a 37°C cell culture incubator for a period of 1 to 4h to develop the fluorescent resorufin product.
- Record the fluorescence of resorufin using a multiwell plate-reading fluorometer fitted with a filter set for 560-nm excitation and 590-nm emission wavelengths. (Note: The spectra of resorufin will enable a variety of filter sets to be used. Data can be collected using excitation filters in the range of 530-570nm and emission filters in the range of 580-620 nm.)
- Plot fluorescence vs cell number.
Each assay format has its own set of advantages and disadvantages. The tetrazolium assay is currently the most widely used method of estimating the number of viable cells in multiwell plates and is the most often cited in the scientific literature. The resazurin reduction assay is functionally similar to the tetrazolium assay, except it has the optional advantage of using fluorescent detection methods. In contrast to the tetrazolium and resazurin reduction assays that require 1- 4h of incubation to obtain meaningful results, data from ATP and LDH assays can be obtained after a 10-min incubation. The ATP and LDH assays lyse cells and thus provide "a snapshot" of the condition of the cells at time of lysis. This advantage provides a quicker assay and avoids any toxic effects of the assay reagents that may occur during the incubation period. For many applications, the ATP assay may be the best choice. It is the most sensitive, provides results faster than any of the other assays, and is the easiest to use. However, one of the limitations of the ATP assay is that it requires a multiwell plate-reading luminometer that may not be available in all laboratories. The choice of which particular assay format to use may depend on the availability of instruments to record data, the detection sensitivity required, the number of samples to be measured, and whether total cell number, viable cell number, or nonviable cell number is chosen as an end point for measurement.
Multiplexing of two assays to gather more than one type of data from the same experimental well may help eliminate the possibility of artifacts. For example, an LDH cytotoxicity assay and an ATP viability assay can be done using the same sample of cells. A small aliquot of culture supernatant can be used for estimating the number of dead cells by measuring the amount of LDH released into the culture medium. Because the sample of cells remains intact, an ATP assay (or any of the other methods) can be used to measure viable or total cell number.
Temperature is a factor that affects the performance of the aforementioned assays because of its effect on enzymatic rates. It is critical to run the assays at a uniform temperature to ensure reproducibility across a single plate or among stacks of several plates. For assays developed at room temperature, it is important to ensure adequate equilibration of samples after the removal of assay plates from a 37°C incubator to avoid differential temperature gradients resulting in "edge effects." Stacking large numbers of assay plates together in close proximity should be avoided to ensure complete temperature equilibration.
Proper negative and positive controls are required to test whether compounds being measured have an effect on the assay chemistry or result in artifacts. For example, strong reducing compounds may interfere with procedures using redox dyes such as the tetrazolium or resazurin reduction assays. Culture medium supplemented with pyruvate will slow the rate of the LDH reaction and thus will require longer incubation periods to generate an adequate fluorescent signal in the CytoTox-ONE assay. In addition, different animal sera have different amounts of LDH activity that will influence background fluorescence. To correct for many of these factors, use of a "no treatment" negative control and a positive control to show maximum effect on each multiwell plate is recommended for all assays.
References
CellTiter 96 AQueous One Solution Cell Proliferation Assay Technical Bulletin #TB245, Promega Corporation.
CellTiter-Blue Cell Viability Assay Technical Bulletin #TB317, Promega Corporation.
CellTiter-Glo Luminescent Cell Viability Assay Technical Bulletin #TB288, Promega Corporation.
Crouch, S. P., et al. (1993). The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 160, 81-88.
CytoTox-ONE Homogeneous Membrane Integrity Assay Technical Bulletin #TB306, Promega Corporation.
Decker, T., and Lohmann-Matthes, M. L. (1988). A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 115, 61-69.
Ekwall, B., et al. (2000). MEIC Evaluation of acute systemic toxicity. VIII. Multivariat partial least squares evaluation, including the selection of a battery of cell line tests with a good prediction of human acute lethal peak blood concentrations for 50 chemicals. Altern. Lab. Anim. 28(Suppl. 1), 201-234.
Gonzales, R. J., and Tarloff, J. B. (2001). Evaluation of hepatic subcellular fractions for Alamar Blue and MTT reductase activity. Toxicol In Vitro 15, 257-259.
Korzeniewski, C., and Callewaert, D. M. (1983). An enzyme-release assay for natural cytotoxicity. J. Immunol. Methods 64, 313-320.
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assay. J. Immunol. Methods 65, 55-63.
O'Brien, J., et al. (2000). Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 267, 5421-5426.
Petty, R. D., et al. (1995). Comparison of MTT and ATP-based assays for measurement of viable cell number. J. Biolumin. Chemilumin. 10, 29-34.
Stanley, P. E. (1986).Extraction of adenosine triphosphate from microbial and somatic cells. Methods Enzymol. 133, 14-22.
Zhang, H.-U., et al. (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67-73.
Support our developers




