Electron Microscopy of Extracted Cytoskeletons: Negative Staining, Cryoelectron Microscopy, and Correlation with Light Microscopy
Our understanding of the function of the cytoskeleton and its associated proteins has been much advanced by developments in imaging methods for light microscopy. With the advent of probes, such as green fluorescent protein, the localisation of gene products in living cells under different physiological conditions is now readily feasible (e.g., Vignal and Resch, 2003). However, to extend our understanding of the functional interactions taking place, parallel information about cellular ultrastructure is essential. Here, electron microscopy (EM) is required to provide information about cell architecture at the nanometre level.
Three major filament systems make up the cytoskeleton: actin filaments, microtubules, and intermediate filaments. When not in organised bundles, actin filaments are poorly preserved after dehydration and embedding in plastic. This fact, taken together with the open, three-dimensional nature of the cytoskeleton networks, has limited the information obtainable by conventional EM methods based on thin sectioning. The methods of choice for studying the cytoskeleton of cultured cells have therefore centered on "whole mount" procedures, involving the culturing of cells on plastic films or coverslips and the preparation of entire cytoskeletons, or their replicas, for electron microscopy. Methods that deliver useful structural information about cytoskeleton organisation include quick freezing and deep etching (Heuser and Kirschner, 1980), critical point drying followed by metal shadowing (Svitkina et al., 1995), and negative staining (reviewed in Small, 1988; Small et al., 1999). More recently, advances in cryoelectron microscopy have opened the way to the study of cytoskeletal structures under conditions that obviate processing steps that could produce artefactual distortions of filament networks (Resch et al., 2002; Medalia et al., 2002). In particular, these latter approaches, involving cryoelectron tomography, promise to deliver new information about ultrastructural interactions in three-dimensional space unobtainable by other means.
II. MATERIALS AND INSTRUMENTATION
A. General
Chloroform (Cat. No. 1.02445.1000), disodium hydrogen phosphate (Cat. No. 1.06580.1000), ethanol (Cat. No. 1.00983.2511), glucose (Cat. No. 1.04074.1000), sodium chloride (Cat. No. 1.06404.1000), and sodium dihydrogen phosphate (Cat. No. 1.06345.1000) are from Merck EGTA (Cat. No. E3889), MES (Cat. No. M2933), taxol ("paclitaxel," Cat. No. T7402), and Triton X-100 (Cat. No. X100) are from Sigma. Phalloidin was a generous gift from Professor H. Faulstich (Heidelberg) and is obtainable from Sigma (Cat. No. P2141). EM-grade glutaraldehyde (Cat. No. R1020) and Formvar (Cat. No. R1201) are from Agar Scientific, Stanstead, UK; nickel 200 mesh hexagonal or nickel H2 finder grids are from Graticules Ltd., Tonbridge, UK; and steel and titanium forceps (number 5) are from Dumont & Fils., Switzerland. Thirteen-millimeter-diameter plastic coverslips (Cat. No. 174950) are from Nunc, Rochester. Further materials required include Dow Corning siliconebased high vaccum grease and Whatman Qualitative No. 1 filter paper. The following are from local suppliers: glassware (50-ml bottle, measuring cylinder, a glass through minimum 7cm deep, a rod or pipette), object slides with frosted ends, Parafilm, transfer pipets, scalpels, and blades. Additionally, cell culture equipment (media, incubators, inverted phase contrast microscope), a basic workshop setup (for producing coverslips with holes), and a routine transmission electron microscope are required.
Sodium silicotungstate (Cat. No. R1230) is from Agar Scientific, Stanstead, UK. Minisart 0.2-µm filters are from Sartorius.
C. Cryo.EM
1,2-Dichlorethane (Cat. No. 8.22346.2500) and methanol (Cat. No. 1.06009.2511) are from Merck, and 87% glycerol (Cat. No. G7757) is from Sigma. Additional laboratory equipment required for producing holey films is a microprobe sonicator and a vacuum evaporator for carbon coating (e.g., Edwards E306).
A freeze-plunging device, either home made (drawings and materials list available upon request) or a commercial apparatus housed in a chemical extractor hood, is needed. At the base of the plunger is a polystyrene box for liquid nitrogen and into this sits a small container for condensing ethane (see Fig. 3). Frost-free liquid nitrogen, gaseous ethane, and a regulator valve with a length of silicon tubing, 4mm inner diameter and 6 mm outer diameter (Tygon 3603, Norton Performance Plastics Corporation, Akron, OH), with one end fitted with a pipette tip with its end cut at a 45° angle, are needed. Note: Remove any sources of ignition from the working area as ethane is highly flammable and explosive. Safety glasses for eye protection or a Plexiglas full face shield, which additionally helps prevent inadvertent breathing onto the cold surfaces, a cause of ice contamination, are required.
Use Whatman qualitative No. 1 filter paper (Cat. No. 1001055), cut to rectangular strips of approximately 1 × 4cm. Grid box (Agar Scientific, Cat. No. G276A) cut into small squares of 15 mm so that there are four grid holes arranged symmetrically within the square, fitted with a clear plastic lid, 13mm circular diameter and 1mm thick, with a 4-mm square notch cut out to allow access to one grid hole at a time is required. This is fitted with metal or nylon screw attached through the center to the grid box base so it can be rotated from one hole to the next and fixed tight for storage. Obtain 50-ml polypropylene conical tube with two approximately 8-mm-diameter holes in the lid to allow nitrogen to enter the tube and for retrieval using long tweezers. Small, portable industrial quality vacuum dewars (do not use houshold vacuum flasks as they can dangerously implode) or a polystyrene container for transporting grids in the grid box is needed. Purchase a large vacuum dewar for long-term storage; it is useful to have a dewar that has numbered and colour-coded holders of the correct size to hold storage tubes.
For electron microscopy, a transmission electron microscope fitted with cryo blades to protect the frozen grid from ice contamination, as well as a cryo holder, cryo transfer station, and temperature controller (Gatan, Pleasanton, USA), are needed.
III. PROCEDURES
A. Negative Staining
1. Preparation of Formvar-Coated Grids
Thin plastic films on grid supports, as used for conventional EM, can be used as a growth substrate for cultured cells. For alternative protocols, see Small and Herzog (1994) and Small and Sechi (1998).
Dissolve 0.4 g Formvar in 50ml chloroform and stir vigorously in a tightly closed bottle for at least 1 h. To produce films without holes, this solution and all glassware used should be kept as dry as possible.
Steps
- Clean an object slide with a frosted end first with distilled water and then with 70% ethanol and allow to dry completely.
- Transfer the Formvar solution into a measuring cylinder wide enough for the slide and submerge the slide, holding it on the frosted end by a clamp or peg attached to a thread, to just below the frosted area. After 30 s, lift the slide smoothly out of the solution and let it dry for 3 min hanging just above the solution surface.
- Drain the excess solution at the bottom of the slide with filter paper and cut the film at the bottom, the left and the right edges, around 1-2 mm inside the edge of the slide. (Do the same for the back side before wetting the film, if both films are to be floated off together.)
- Prepare a large vessel filled to the lip with distilled water, clean the surface with a glass rod from dust, and float off either one or both of the films simultaneously at an angle of 45° resp. 90° onto the surface. A black background below the vessel improves visualisation for this and the following steps. (Note: If difficulties are encountered with floating off the film, try another brand of slides or a different washing protocol; see also Small and Herzog, 1994.)
- Place 200 mesh Ni grids with the dull side downwards onto the film. For easy handling of the magnetic nickel grids, the use of titanium forceps is recommended. Retrieve the film from the water surface with a piece of Parafilm that is put on top of the floating grids/film assembly and then lifted up carefully from one end. (An alternative way to do this is to use a microscope slide: with an address sticker adhered to one side and trimmed at the the edges, hold the slide at a 45° angle (paper side down), touch the film a few millimetres within one of the narrow ends, and slowly submerge the slide into the water. The film with the grids will follow the slide and stick very well.)
- Remove excess water by blotting carefully with filter paper and allow the filmed grids to dry completely. To separate them without damaging the film, cut the film around each individual grid with the tip of a pair of forceps.
Cells are plated onto the grids the same as for coverslips, and the density is chosen to give one or two cells per grid square after attachment and spreading. In general, cells spread more slowly on plastic film than on glass; to encourage attachment and spreading, the filmed grids may be coated with matrix molecules or serum (depending on cell type) prior to plating. Check for adequate cell spreading in an inverted microscope with phase-contrast optics prior to further processing.
In order to render the film more stable in the electron beam, an additional layer of carbon deposited onto the grid can be used. However, under these conditions, we experience a decrease in quality of the negative stain so that we usually apply a thin carbon layer after staining (see later).
To avoid the problem of loose, floating grids, it is an advantage to immobilise them in one way or another (see also Small and Sechi, 1998). To immobilise single grids, we sandwich them between a plastic coverslip with a 2.5- to 2.7-mm hole in the center of a petri dish. Dots of vacuum grease are used to fix the plastic coverslip to the petri dish.
- Prepare plastic coverslips >12mm diameter with a central hole of 2.5-2.7 mm; this is best done by fixing a stack of them tightly in a holder prepared separately for this purpose (Fig. 2a) before drilling. Clean the coverslips by washing them twice with 70% EtOH and once with water, both of them heated in a microwave oven until boiling.
- For cell culture over periods of more than 12h, sterilise grid sets under UV light in an open petri dish for 5 rain. Air bubbles get trapped easily in the mesh, which makes observation of the spreading cells (e.g., in phase contast) difficult. This can be avoided by incubating the grids in sterile water in the fridge overnight before use. This step should be performed individually for each grid in grid boxes (volume per slot approximately 15µl) to avoid the grids adhering to each other.
- Transfer the grids, with the filmed side up, to sterile (and dry) petri dishes and fasten them with the plastic coverslips: Apply small spots of high vacuum grease onto the edge of the coverslips and press them down into the petri dishes, with the grid trapped below the hole. Only coverslips with a regular hole and fiat edge should be used.
- After they are mounted, the films can be protein coated and cell culture can be carried out as usual.
3. Extraction/Fixation
Solutions
- Prewarmed phosphate-buffered Saline (PBS, 150mM NaCl, 3 mM NaH2PO4, 8 mM Na2HPO4, pH 7.4)
- Prewarmed extraction buffer [0.25% Triton X-100 (diluted from a 20% stock) and 0.5% EM-grade glutaraldehyde (diluted from a 25% stock) in CB: 10 mM MES, 150 mM NaCl, 5 mM EGTA, 5 mM MgCl2, 5 mM glucose, pH 6.1]
- Fixation buffer at room temperature (1.0% EMgrade glutaraldehyde from a 25% stock in CB)
- Phalloidin, 1mg/ml in MeOH, stored at -20°C
- Taxol, 10 mM in dimethyl sulfoxide, stored at -20°C
- Remove cover glasses and transfer grids to a small petridish with prewarmed PBS for a brief wash. To avoid bending in the transfer step, a magnetised steel forceps is useful for initially lifting the grid so that it can be grabbed more easily with another pair of (titanium) forceps.
- Aspirate the PBS carefully, do not allow to dry, and replace it with prewarmed extraction buffer; incubate for lmin.
- Replace the extraction buffer with fixation buffer and fix for >20min up to overnight at 4°C; to improve the preservation of actin and microtubules, 10µg/ml phalloidin or 10µg/ml taxol can be added.
- Negative Staining and Electron Microscopy
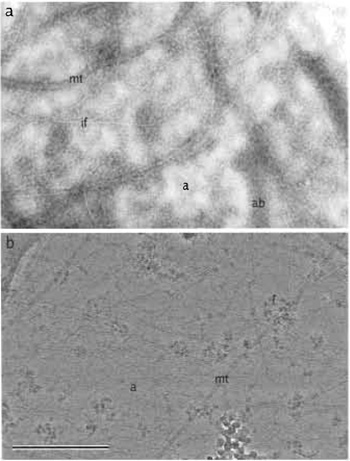
FIGURE 1 Extracted cytoskeletal networks in thin regions of cells visualised by (a) negative staining with sodium silicotungstate and (b) cryoelectron microscopy, a, actin; ab, actin bundle; mt, microtubule; if, intermediate filaments; r, ribosome aggregation. Bar: 500nm.

2% sodium silicotungstate in ddH2O, adjusted to
pH 7 with NaOH (equilibration of pH can take a long time) and filtered through a 0.20-µm filter.
Steps
- Clamp the grid in a pair of forceps using a large paper clip over the shaft to hold the tips together; do not allow the sample to dry at this step!
- Rinse with a few drops of the negative stain from a transfer pipette. After a few seconds, drain the stain from the entire back side of the grid with filter paper to remove it as completely as possible. On the front, drain only from the edges to leave a thin film so as not to damage the sample. Special care has also to be taken to drain excess stain from between the tips of the forceps: Bring the edge of the filter paper into contact with the edge of the grid at the contact point with the forceps.
- Allow to air dry and observe in a routine TEM (Fig. 1a). To avoid drift of the plastic film upon exposure to the electron beam, a thin layer of carbon can be deposited onto the film in a vacuum evaporator.
B. Cryo-EM
This section describes the method from Resch et al. (2002) in more detail.
For unstained specimens, the support film itself contributes a significant background noise in the electron microscope image. To improve contrast conditions in the cryo-EM, we have therefore used holey films to support cells and have selected areas for structural analysis where parts of cells span the holes. The method for making holey films was derived from that described in Hodgkinson and Steffen (2001); it was optimised for a hole/film ratio that was significantly lower than for films used for molecular suspensions to allow the cells to attach and spread.
Solution
Dissolve 0.25 g Formvar in 50 ml chloroform and stir in a tightly closed bottle for at least 1 h. After the Formvar is dissolved, add 150µl of 50% glycerol in water; shake the solution vigorously for 1 min.
Steps
 |
| FIGURE 2 (a) Device used for mounting the plastic coverslips for drilling consists of a plastic body (1) with an elongated grove (here horizontal) and a central round indentation (2) in which a stack of cover glasses sit. They are mounted by a Plexiglas bar (3) on top of the groove, which is pressed down firmly onto them by two screws at its end (4). Drilling is done via a central hole (5) in the Plexiglas bar. (b) Perforated carbon film as typically used for cultivating cells for cryo-EM on a 200 mesh hexagonal Ni grid. (c) Two setups to mount grids for correlative LM/EM in petri dishes with a coverslip inset in the bottom on an inverted microscope; samples are covered with medium, the objective is coming from below. (Left) Upright setup: The grid with cells up (1) is mounted by a plastic coverslip with a central hole (2) that is attached via high vaccuum grease (3). (Right) Inverted setup for fluorescence microscopy: The grid with cells on it facing downwards (1) is put on top of two other nickel grids (4) that serve as spacers for the cells and fixed with the coverslip as described earlier. |
- Prepare a clean object slide as described previously.
- Sonicate the Formvar/glycerol solution for 3 min.
- Rapidly transfer the solution to a staining jar, dip the slide into it, remove, and allow to air dry for a few minutes.
- Float off both sides of the film as described earlier and place 200 mesh Ni grids with the dull side facing the film on top. In this case, the grids should not be placed too close together on the film, as the filter paper used to retrieve the grids will not adhere sufficiently!
- Retrieve the grids and the film with a piece of filter paper, not with Parafilm or a slide, and allow to dry.
- Prepare a stack of filter paper in a large glass petri dish saturated with MeOH and incubate the grid set on this, with the film up, for 10 min. This serves to perforate the pseudoholes. Remove and allow to air dry completely before proceeding.
- At this stage, we recommend checking the film with a phase-contrast microscope for the size and distribution of holes, which is affected by the amount of glycerol as well as by the power and duration of the sonication. If neccessary, vary these factors.
- Coat the grids with a thick (dark grey) layer of carbon in a high vacuum evaporator.
- Prepare a bed of filter paper saturated with 1,2- dichlorethane. Place the set of grids, film side up, onto the filter paper and leave for 2 h to dissolve the plastic film. If the filter paper bends up at the edges, either cut it into smaller pieces or use small pieces of glass to hold it down.
- After final drying, check a few grids in the EM (see Fig. 2b) and store them individually in grid boxes.
2. Cell Culture
For preparation of cells for cryo-EM, the same procedure is used as for negative staining. However, making the cells attach and spread on perforated films might require additional steps, including coating with matrix molecules (for general strategies, see also Resch et al., 2002; and Vignal and Resch, 2003).
3. Extraction/Fixation
For cryo-EM as outlined here, removal of the membrane and cytosolic protein is necessary to visualise the cytoskeletal networks and actin filament subunit structure clearly. It is also necessary to fix the cytoskeletons lightly during this extraction process to stabilise the networks. Medalia et al. (2002) have applied cryoelectron tomography to unextracted cells, in which case filament networks are visible only after complex image-processing regimes.
Here, the extraction/fixation protocol is the same as for negative staining, except that postfixation seems not to be neccessary.
4. Cryo Plunging of Specimen Grids
The objective of plunge freezing is to obtain a well distributed, quick frozen specimen in a homogeneously thin layer of vitrified ice uncontaminated with frost. For this, the blotting regime is critical. The amount of blotting required can be sample/buffer dependent so this might have to be adapted for different preparations. It is important to remain consistent but because in some cases even subsecond differences in blotting times can cause variability with ice thickness, differences between grids are unavoidable. In order to counter this, make several grids at each session.
If a washing buffer is to be used, it is important that it is at the same osmolarity as the cell culture media, as the integrity of the cells could be compromised.
Steps
 |
| FIGURE 3 Working setup as typically used for freeze plunging. (1) Gaseous ethane bottle, (2) regulator valve with Tygon tubing and pipette tip, (3) full face shield, (4) filter paper strips, (5) polystyrene box for liquid nitrogen, (6) small container (sitting on an aluminum block) for condensing ethane, (7) plunger arm, fixed with a pair of forceps holding the grid (arrowhead), (8) 50-ml conical tube for grid box storage, (9) tools (fine and large forceps, screwdrivers), and (10) sample transport dewar. For details, see text. |
- Set up your workspace with all the required tools close at hand prior to freezing (Fig. 3). It is important to have good light and it is useful to have additional light (e.g., equipoise lamp) that can be positioned facing toward the grid for observing the blotting step when plunging. Frost in the liquid nitrogen and ethane can be a source of contamination on your grids so always try to minimise exposure to water vapour. Always keep the nitrogen containers sealed with lids and do not breathe onto the nitrogen or ethane surfaces. Precool all tools that come into contact with the grid or grid box, as they may heat the specimen, causing cubic or hexagonal ice to form.
- Set up the plunger. Check the alignment of the forceps in the plunging arm so that the forceps will plunge the grid a few millimetres below the surface of the liquified ethane. Do not adjust the drop height so that the grid gets too near the base of the ethane container as ethane ice forms there, which will damage the grid if it comes into contact with it.
- Put on eye protection or a face shield and fill the polystyrene container with liquid nitrogen. Also fill the portable transfer dewar with liquid nitrogen and submerge the polypropylene conical tube into this. Keep a lid on the transfer dewar to minimise contamination.
- Precool the small ethane container with liquid nitrogen. Make sure all liquid nitrogen is removed from the inside of the container as it interferes with the liquifying of the ethane. Place one of the grid storage boxes into the liquid nitrogen.
- Place the pipette tip from the ethane supply into the bottom of the small ethane container and slowly open the regulator valve. After a short time, the ethane gas will start to condense on the bottom of the container. This is normally accompanied by a high-pitched squeal. Keeping the pipette tip just below the surface of the liquid ethane and gently moving it around the inner walls allow the level to rise to the top of the ethane container. Keep the tip moving in the ethane so that the tip does not ice up and block. Do not allow the liquid ethane to spill over into the nitrogen. You can use a double-walled ethane container with a small gap between the walls to avoid this. Note: Liquid ethane can cause serious burns if it comes into contact with your skin or eyes so always exercise caution and regulate the ethane to a slow and gentle flow.
- After a short time, the liquified ethane will start to solidify. If this happens, you must deice by inserting the pipette tip into the ethane. Slowly open the ethane gas supply as you do not want to splash ethane into the nitrogen. Gently move the pipette tip around the edges of the ethane container until the ethane liquifies.
- Pick up a grid by the edge, sample side up with forceps. The samples are usually washed briefly, immediately prior to plunging. Cut a piece of Parafilm, place on a flat surface, and apply 2 × 50-µl drops of washing buffer, e.g., prewarmed PBS. Keeping the grid in the tweezers, blot the grid side on with a piece of filter paper. Apply the grid, sample side down to the surface of the washing droplet, blot, and place onto the second drop. Remove the grid; a droplet from the washing buffer should remain on the grid surface. Place the forceps into the clamp of the plunger arm with the sample (and droplet) facing towards you.
-

FIGURE 4 Blotting of the grid immediately prior to freeze plunging: (a) when first touching the grid and droplet with a strip of filter paper, an outline of the grid is clearly visible through the paper and (b) as the liquid is absorbed by the filter paper, the droplet imprint will increase in diameter until most of the liquid is removed from the grid surface. The grid outline will disappear and the paper will pull away from the grid. At this moment, plunge the grid immediately. For more details, see text. Using a strip of the Whatman qualitative No. 1 filter paper, carefully blot the surface of the grid. Use a visual assessment to roughly gauge the degree of blotting (Fig. 4): When first touching the grid, the filter paper will pull onto the droplet by surface tension and you should see the outline image of the grid, through the paper. Once most of the surface liquid is adsorbed by the paper you will reach a point where the grid outline will suddenly disappear and the paper will also pull away. This is the time when the sample should be plunged rapidly into the ethane. Waiting even a few seconds after the pull-away point can result in excess removal of liquid, sample damage, and drying. - While keeping the grid under the surface of the ethane, remove the tweezers from the plunging arm. Raise the plunging arm away from the tweezers. Carefully remove the tweezers and grid from the ethane and immediately hand plunge the grid into the liquid nitrogen. Make sure the grid stays under the cold nitrogen vapour cloud to keep it from warming and frosting. This can be achieved by having sides on the polystyrene liquid nitrogen box that are higher than the top level of the liquid ethane container.
- Transfer the grid, keeping it submerged under the liquid nitrogen, into one of the storage slots in the grid box. After all samples have been loaded, close the lid firmly with a screwdriver. Place the liquid nitrogenfilled transfer dewar containing the polypropylene tube close to the plunger. Using a large pair of tweezers, quickly transfer the grid box from the plunger into the tube. Screw the lid onto the tube.
- Move the tube and grid boxes holding the samples into a long-term storage dewar. As long as the nitrogen is kept topped off the grids can remain here for long periods (sometimes years) without deterioration until you are ready to view them in the microscope. We totally dry our dewars once a year with a stream of dry nitrogen gas to remove any frost that may accumulate with time. Transfer all grids to another frost-free container while drying the original dewar.

Steps
- Using a precooled pair of large tweezers, remove the grid storage box holding the frozen grids and place in a liquid nitrogen-filled transfer dewar.
- At the electron microscope, fill the anticontaminator and cryo-blade dewars.
- If the cryo holder has been pumped (it is recommended to continually vacuum pump the cryo holder dewar when not being used), remove it from the vacuum pump stand. Insert the holder carefully into the loading station. Connect the cable for temperature measurement between the cryo holder and the temperature controller.
- Remove the clip ring from the cryo holder with the clip ring tool.
- Put on eye protection or a face shield.
- Fill the specimen holder dewar and the loading station dewar with liquid nitrogen. After each filling with liquid nitrogen, replace the Plexiglas lid on the loading station dewar to minimise frost contamination. Keep the cap on the holder dewar, as it keeps the outer sides from cooling. Make sure you open and close the tip shutter as you cool as this prevents the shutter from jamming. You will need to keep topping off the level so that nitrogen sits just below the tip of the sample holder. Wait for the temperature controller to read at least -160°C before loading your sample.
- Placing the transfer dewar as close as possible to the specimen holder dewar, rapidly transfer the grid box holding the frozen specimens to the specimen holder dewar. Use a precooled pair of large tweezers.
- Precool a screwdriver and loosen the lid of the grid box.
- Check that the level of the liquid nitrogen is just below the tip of the holder; cool a pair of No. 5 forceps and the clip ring tool in the loading station dewar.
- Using the forceps, remove a grid from the grid box. Making sure you keep the grid within the cold nitrogen vapour, place the grid into the recess in the cryo holder tip. Centre the grid in the recess and fasten with the clip ring. Immediately close the holder tip shutter.
- Replace the Plexiglas lid on the loading station dewar and lift the loading station and cryo holder and place onto the console desk of the microscope. We 15rotect the console surface from nitrogen damage with a metal splash guard (Gowen and Burger, 1998).
- Cycle the roughing pump on the microscope so that the pump lines to the microscope sample stage (e.g., compustage on an FEI machine) are under good vacuum before insertion of the cryo holder.
- Remove the holder from the loading station and carefully insert into the microscope stage for preevacuation. (It can be useful to pretilt the stage to around -60° so that the holder dewar neck is positioned at "3 o'clock" instead of "6 o'clock" to lessen nitrogen spillage when the holder is introduced.) Wait for the vacuum to be restored and then insert the holder all the way into the microscope.
- Allow at least 20min for the temperature to stabilise and the vacuum to recover. Temperature variations cause the holder to drift. Connect the cable from the temperature controller and monitor the temperature. During the transfer the temperature should not warm to above about -150°C. Normally the holder will stabilise at around -180°C. Do not leave the cable connected to the holder as this can also cause drift effects.
To reduce damage when viewing cryo grids in the electron microscope, it is necessary to have low-dose software, which allows you to minimise the electron dose applied to the sample. Always use the beam blanker when you are not observing the specimen. The microscope should be well aligned and set up for lowdose conditions. To lower the electron intensity at the sample, you can use a small condenser aperture, small spot size, and low emission settings.
Steps
- Once the holder temperature has equilibrated and drift minimised, the grid should be screened at low magnification (usually at around 3000x or less) to find promising cell areas for imaging.
- Images can be acquired either digitally using a CCD camera system or taken onto plate film. If required, images on plate film can be scanned digitally using a suitable high-resolution film scanner.
- In some instances, micrographs may appear to have an uneven intensity difference across the image due to local variations in ice thickness. Applying a high-pass filter helps even the intensity, which allows subsequent contrast adjustments to be made more easily (Fig. 1b).
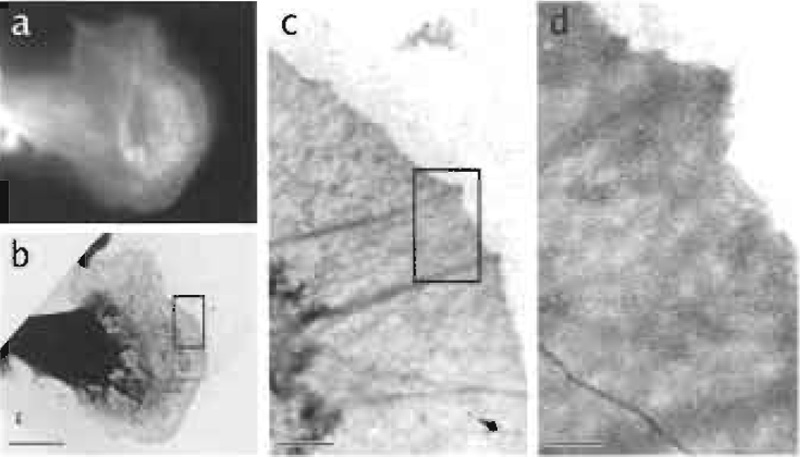 |
| FIGURE 5 Correlative light and electron microscopy as demonstrated by a B16 mouse melanoma cell: (a) Cell stained with fluorescently labelled phalloidin and visualised by epifluorescence microscopy, (b) corresponding view of the same cell afer negative staining, (c and d) higher magnification insets of the previous images. Bars (a and b) = 10 µm, (c) = 1 µm, (d) = 250 nm. |
For situations in which the cell shape cannot be visualised clearly (e.g., frozen hydrated samples) or in which the function cannot be deduced solely from the morphology but only from a marker protein (e.g., GPFVASP for protruding lamellipodia; Rottner et al., 1999), correlative light and electron microscopy on the same cell can be very helpful in establishing the structure-function link as discussed earlier. This correlation should be relatively straightforward for the classical methods, where cells are grown directly on glass coverslips with etched finder patterns; appealing examples can be found in Svitkina and Borisy (1999). For negative staining and cryo-EM, where cells are grown directly on grids, finder grids with indexed grid holes have to be used (Fig. 5).
- Prepare grids with a Formvar or a holey carbon film as described earlier; use H2 Ni finder grids instead of hexagonal 200 mesh grids to be able to relocate your samples.
- Spread cells on these grids. For correlative LM/EM, it is essential to remove any air bubbles trapped in the mesh by preincubating them in water as described previously.
- Mount the grids individually for light microscopy; how exactly this is done depends on the setup and the cell line used. As a general guide, care has to be taken to (1) mount the grid flat (e.g., by using the method described earlier with coverslips with a hole drilled in the center), (2) have the cells as close to the objective to avoid problems with focus, and (3) mount grids with a carbon film upside down for epifluorescence to avoid loss of intensity. Two sample setups using petri dishes with a glass coverslip at the bottom are shown in Fig. 2c.
- For the light optical observation of the cells, there are several possibilities available: (1) living cells in phase contrast, (2) living cells expressing a fluorescent protein (conjugate), and (3) fixed cells after staining with a fluorescent probe. For the latter method, it is essential to minimise the damage of the substructure by prolonged incubation steps in the staining protocol, e.g., fluorescently labelled phalloidin can be included directly in the fixative.
- Record the position of each cell observed in a map of the finder grid; such a map for the H2 grids is provided by the authors at ftp://cellix.imba.oeaw.ac. at/Protocols/findergrids.zip.
- Proceed as described earlier for negative staining or cryo-EM.
Acknowledgments
The authors thank Johanna Prast, Institute of Molecular Biology, Salzburg, for her continuous support, Marietta Schupp, Photo Lab, EMBL Heidelberg, for her contributions (Figs. 3 and 4), and Johann Diendorfer, Institute of Molecular Biology, Salzburg, for his help with producing coverslips with holes. GPR is supported by project P-15710 of the Austrian Science Foundation.
Gowen, B. E., and Burger, L. (1998). Cryo-TEM liquid nitrogen splash guard. J. Micros. 191, 320-322.
Heuser, J. E., and Kirschner, M. W. (1980). Filament organization revealed in platinum replicas of freeze-dried cytoskeletons. J. Cell Biol. 86, 212-234.
Hodgkinson, J. L., and Steffen, W. (2001). Direct labeling of components in protein complexes by immuno-electron microscopy. Methods Mol. Biol. 161, 133-139.
Medalia, O., Weber, I., Frangakis, A. S., Nicastro, D., Gerisch, G., and Baumeister, W. (2002). Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science 298, 1155-1157.
Resch, G. P., Goldie, K. N., Krebs, A., Hoenger, A., and Small, J. V. (2002). Visualisation of the actin cytoskeleton by cryo-electron microscopy. J. Cell Sci. 115, 1877-1882.
Rottner, K., Behrendt, B., Small, J. V., and Wehland, J. (1999). VASP dynamics during lamellipodia protrusion. Nature Ceil Biol. 1, 321-322.
Small, J. V. (1988). The actin cytoskeleton. Electr Microsc. Rev. 1, 155-174.
Small, J. V., and Herzog, M. (1994). Whole-mount electron microscopy of the cytoskeleton: Negative staining methods. In "Cell Biology: A Laboratory Handbook" (J. E. Celis, ed.), Vol. 2, pp. 135-139. Academic Press, San Diego.
Small, J. V., and Sechi, A. (1998). Whole-mount electron microscopy of the cytoskeleton: Negative staining methods. In "Cell Biology: A Laboratory Handbook" (J. E. Celis, ed.), 2nd ed., Vol. 3, pp. 285-291. Academic Press, San Diego.
Svitkina, T. M., and Borisy, G. G. (1999). Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament arrays in lamellipodia. J. Cell Biol. 145, 1009-1026.
Svitkina, T. M., Verkhovsky, A. B., and Borisy, G. G. (1995). Improved procedure for electron microscopic visualization of the cytoskeleton of cultured cells. J. Struct. Biol. 115, 290-303.
Vignal, E., and Resch, G. P. (2003). Shedding Light and Electrons on the Lamellipodium: Imaging the Motor of Crawling Cells. Biotechniques 34(4), 780-789.
Support our developers




