Immortalization of Primary Human Cells with Telomerase
One major obstacle to the immortalization of primary human cells is telomere-controlled senescence. Telomere-controlled senescence is caused by the shortening of the telomeres that occurs each time somatic human cells divide. The enzyme telomerase can prevent the erosion of telomeres and block the onset of telomere-controlled senescence, but its expression is restricted to the human embryo and, in the adult, to rare cells of the blood, skin, and digestive track. However, we and others have shown that the transfer of an exogenous hTERT cDNA, encoding the catalytic subunit of human telomerase, can be used to prevent telomere shortening, overcome telomerecontrolled senescence, and immortalize primary human cells (Bodnar et al., 1998). Most importantly, hTERT alone can immortalize primary human cells without causing cancer-associated changes or altering phenotypic properties (Jiang et al., 1999; Morales et al., 1999; Ouellette et al., 2000). Primary human cells that have been immortalized successfully with hTERT alone include fibroblasts, retinal pigmented epithelial cells, endothelial cells, myometrial cells, esophageal squamous cells, mammary epithelial cells, keratinocytes, osteoblasts, and Nestin-positive cells of the pancreas (Bodnar et al., 1998; Yang et al., 1999; Ramirez et al., 2001; Yudoh et al., 2001; Condon et al., 2002; Lee et al., 2003; Morales et al., 2003). This article describes the use of hTERT for the purpose of immortalizing primary human cells using the primary human fibroblast as an example.
Packaging cell line Phoenix-ampho (ΦNX-A) for the production of amphotropic viruses is available from Dr. Gary Nolan (Stanford University, Stanford, CA; http://www.stanford.edu/group/nolan/). Retroviral vectors pBabePuro and pBabePuro-hTERT are from Geron Corp. (Menlo Park, CA; Ouellette et al., 1999). Dulbecco's modified Eagle's medium (DMEM; Cat. No. 11995), medium 199 (Cat. No. 11150), gentamicin (Cat. No. 15710), and trypsin-EDTA (0.05% trypsin, 0.53 mM EDTA; Cat. No. 25300) are from Invitrogen Corp. (Carlsbad, CA). Electrophoresis apparatus (Cat. Series 21070), including glass plates, spacers, and combs, are from Life Technologies (Rockville, MD). Cosmic calf serum (Cat. No. SH30087) and fetal bovine serum (Cat. No. SH30070) are from HyClone (Logan, UT). The polysulfone filter (0.45µm, Cat. No. DD50402550) is from Life Science Products Inc. (Frederick, CO). Dimethyl sulfoxide (DMSO; Cat. No. D 2650), polybrene (Cat. No. H 9268), puromycin (Cat. No. P 7255), and bovine serum albumin (BSA; Cat. No. A 2153) are from Sigma-Aldrich (St. Louis, MO). The MBS mammalian CaPO4 transfection kit is from Stratagene (Cat. No. 200388; La Jolla, CA). The TrapEZE kit is from Serologicals Corp. (Cat. No. S7700; Norcross, GA). N,N,N',N'-Tetramethylenediamine (TEMED; Cat. No. 161-0800) and 40% acrylamide:bisacrylamide [19:1] solution (Cat. No. 161-0146) are from Bio-Rad (Hercules, CA). The PhosphorImager is from Molecular Dynamics (Model No. 810; Sunnyvale, CA). Nalgene Cryo 1°C freezing containers are from Nalge Nunc International (Cat. No. 5100-0001; Rochester, NY). T4 polynucleotide kinase is from New-England BioLabs (Cat. No. M0201S; Beverly, MA). [γ-32P]ATP is from New England Nuclear (Cat. No. 502A12070; Boston, MA). The polymerase chain reaction (PCR) is performed using a PCRExpress thermocycler from Hybaid (Ashford, Middlesex, UK). All other chemicals are from Fisher Biotechnology (Fait Lawn, NJ), Fisher Scientific Co. (Pittsburgh, PA), or Sigma-Aldrich (St. Louis, MO).
A. Production of Replication-Defective Retroviruses Carrying the hTERT cDNA
Primary human cells tend to transfect very poorly. Consequently, the preferred method of transferring exogenous telomerase to such cells is their transduction with replication-defective retroviruses carrying an hTERT cDNA. The following procedure, modified from Pear et al. (1993), yields high-titer viruses following the transient transfection of ΦNX-A cells, a retroviral packaging cell line.
Solutions
- 293T medium: To 500 µl of DMEM, add 50 ml of fetal bovine serum and 500µl of 10mg/ml gentamicin. Store at 4°C and protect from light.
- Complete medium X: To a clean sterile 500-ml bottle, combine 400 ml of DMEM, 100 ml of medium 199, 50ml of cosmic calf serum, and 500µl of 10mg/ml gentamicin. Store at 4°C and protect from light.
- In a 37°C water bath, thaw a vial of ΦNX-A cells and transfer cells to a culture dish containing 293T medium. Incubate cells at 37°C under 5% CO2. The next day, replace medium with fresh medium.
- Expand ΦNX-A cells in 293T medium using split ratios of I : 4 to 1:6. Avoid letting the cells grow beyond 90% confluence. To split cells, remove medium by aspiration, gently wash cells once with PBS. Take extra care not to dislodge the cells, as ΦNX-A cells tend to detach very easily. Add 2ml of trypsin-EDTA per 100-mm dish and let cells detach at 37°C for 1-2min. Tap dish to help dislodge cells, add 5 ml of 293T medium, and pipette up and down to break cell clusters. Transfer suspension to a sterile tube and pellet cells by lowspeed centrifugation (2000g for 5 min at room temperature). Remove supernatant and resuspend cells in 5 ml of 293T medium.
- Following the manufacturer's instructions, count cells using a Coulter counter or hemacytometer. In two 100-mm plates containing 293T medium, seed 5.0 × 106 cells/dish. Freeze remaining cells to replenish archival stocks, if needed (refer to Section III,C).
- On the following day, ΦNX-A cells should be ~80% confluent and ready to be transfected with the retroviral vectors. Transfect each dish with 10-20µg of plasmid; one with vector pBabePuro-hTERT and the other with the empty vector, pBabePuro. ΦNX-A cells are transfected most easily using the CaPO4 method, but other methods can be used as well. To CaPO4 transfect the ΦNX-A cells, we have used the Stratagene's MBS mammalian transfection kit following the manufacturer's instructions. In a 5-ml Falcon 2054 polystyrene tube, prepare a calcium-DNA precipitate by mixing plasmid DNA (10-20 µg in 450 µl of sterile water) with 50µl of solution I (2.5M CaCl2) and then adding 500µl of solution II [N,N-bis(2- hydroxyethyl)-2-aminoethanesulfonic acid in buffered saline].
- Incubate the calcium-DNA mixtures at room temperature for 10-20min. In the meantime, prepare the cells by washing cells once with PBS and feeding them with 10ml/dish of 293T medium, in which 6% modified bovine serum (provided by the kit) is used in place of the fetal bovine serum.
- Resuspend the DNA precipitate gently and then apply dropwise in a circular motion to the dishes, as to distribute the DNA evenly. Swirl dishes once.
- Incubate cells at 37°C under 5% CO2 for 3 h.
- Remove medium by aspiration.
- Wash cells once with sterile PBS and feed with 293T medium. Return cells to 37°C under 5% CO2. Cells have now been transfected and should start producing retroviruses within the next 24 h.
- Viruses can be collected in three consecutive harvests over the next 24-72h posttransfection. To collect viruses, replace the 293T medium with 4-5 ml of the target cells culture medium, in which the viruses will now be allowed to accumulate. For primary human fibroblasts, collect viruses in complete medium X. Return cells to 37°C under 5% CO2 with the dishes spread on a leveled shelf to ensure good coverage of all surfaces.
- After 8-16h of exposure to the cells, harvest supernatants and then force through a 0.45-µm polysulfone filter so that all remaining cells are removed. Bleach and discard the transfected cells once the last batch of viral supernatant has been collected.
- Keep viral supernatants either on ice and use within the hour or else aliquot, freeze, and store at -80°C where they can last for approximately 6 months. Label each aliquot with name of retroviral vector, harvest medium, date, and harvest number (1st, 2nd, etc...). Freezing does not appear to cause substantial drops in titer, but cycles of freeze/thaw do.
Solutions
- 400µg/ml polybrene: Dissolve 8 mg of polybrene in 20ml of deionized double-distilled water. Sterilize by filtration through a 0.45-µm polysulfone filter. Store at 4°C.
- 500µg/ml puromycin: Dissolve 10rag of puromycin in 20ml of deionized double-distilled water. Sterilize by filtration through a 0.45-µm polysulfone filter. Store aliquots at-20°C.
Steps
- Thaw primary human fibroblasts in complete medium X. Incubate cells at 37°C under 5% CO2. The next day, replace medium with fresh medium.
- Expand cells in complete medium X using split ratios of 1:2, 1:4, and 1:8 for late, mid, and early passage cells, respectively. Avoid leaving monolayers at full confluence for extensive periods of time, as cells may then resist trypsinization. To split cells, remove medium and wash cells twice with 2ml of trypsin-EDTA per 100-mm dish. Incubate dishes at 37°C until cells have detached (1-3 min). Tap dishes to help dislodge cells, add 5 ml of complete medium X, and pipette up and down to dissociate clumps. Transfer suspension to a sterile tube and pellet cells by low-speed centrifugation (2000g for 5min at room temperature). Remove supernatant and resuspend cells in 5ml of complete medium X. While keeping track of population doublings (see later), expand fibroblasts until sufficient cells are available.
- Seed four wells of a 6-well plate with target cells so that the cells are in log-phase growth on the next day. For primary human fibroblasts, seed cells in complete medium X at 2 × 105 cells per well. Take note of the population doubling of the seeded cells as the initial PD (or PDi). Freeze remaining cells to replenish archival stocks, if needed. Label frozen vials with name of strain, date, population doubling, and approximate number of cells per vial (refer to Section III,C).
- Incubate dishes overnight at 37°C under 5% CO2.
- Remove the medium from all dishes. Infect one dish with viruses carrying hTERT (sample A; infected with pBabePuro-hTERT) and a second dish with control viruses carrying no insert (sample B; infected with pBabePuro). Feed the remaining two dishes with virus-free medium (samples C and D; uninfected). Perform infections at 37°C under 5% CO2 for 8-16h using 1-2 ml of a mixture containing I volume of viral supernatant, 1 volume of the target cells culture medium, and 4~tg/ml polybrene. Within 24-48 h, cells can be infected sequentially with all of the different harvests of the same virus.
- Remove the last viral supernatants and replenish all four dishes with the target cell culture medium, using complete medium X for primary human fibroblasts. Return cells at 37°C under 5% CO2.
- Let cells divide once or twice over the next 24- 48 h to allow integration of the viral genomes.
- Select cells for viral integrations using puromycin. The exact dose of puromycin to use should be determined in a preliminary experiment, in which uninfected cells are exposed to increasing doses of puromycin (e.g., 0, 250, 500, 750, 1000ng/ml) for 7-10 days; use the lowest dose that kills all cells. For primary human fibroblasts, 500-750ng/ml is a good starting point. Add puromycin to sample A, B, and C. Leave sample D puromycin free.
- Maintain cells in continuous log-phase growth. Samples that reach confluence should be trypsinized and expanded to a larger dish; do not discard any excess cells yet. Replace puromycin-containing media as needed or twice a week until selection is complete. Selection is complete when all of the uninfected cells of control sample C have died, after typically 7-10 days of selection.
- Expand cells until samples A and B have reached the size of a 100-mm dish. Count total number of cells in samples A, B, and D. Evaluate the number of population doublings done during the infection/selection phases of the experiment (ΔPDi/s). For this purpose, the simplest approximation is to assume that samples A, B, and D have done an equivalent number of doublings during this same period of time. Using sample D as a reference, calculate ΔPDi/s knowing that ΔPDi/s = log[(final number of cells in sample D) ÷ (initial number of cells plated in step 3)] / log[2]. Discard sample D and set PD of samples A and B to PDtime0 = PDi + ΔPDi/s.
- Cells are now ready to be tested for telomerase activity and assessed for life span extension. Put aside 5 × 104 cells of each sample for a telomerase assay (see Section III,C). Start a growth curve by seeding 2 × 105 cells of each sample in a 100-mm dish (see Section III,C). Freeze remaining cells and label vial with name of strain, vector transduced, population doubling (PDtime0), and approximate number of cells.
C. Other Procedures Related to the Immortalization of Human Primary Fibroblasts
1. Freezing ΦNX-A Cells and Primary Human Cells
Freeze medium: Mix 10ml of DMSO with 90ml of fetal bovine serum. Store aliquots at-20°C and keep working solution at 4°C.
Steps
- Trypsinize cells as described earlier, resuspend in culture medium, and recover by low-speed centrifugation (2000g for 5 min at room temperature).
- Remove supernatant by aspiration, and resuspend pellet in freeze medium so that each milliliter contains the cell equivalent of 25-150cm2 of confluent dish surface.
- Aliquot suspension in 2-ml cryogenic vials, at 1 ml per vial.
- Label vials with name of sample, population doublings, date, and approximate number of cells.
- Place vials into a Nalgene Cryo 1°C freezing container filled with fresh isopropanol. Transfer container to-80°C.
- On the next day, transfer the frozen vials to either a liquid nitrogen tank or a -135°C freezer.
2. Measuring Cellular life Span
To verify that exogenous telomerase has an extended cellular life span, a growth curve is generated to measure and compare the life span of the vector- and hTERT-transduced cells.
- Maintain the vector- and hTERT-transduced cells in continuous log-phase growth as cells are counted once a week. For this purpose, seed cells at a density that requires a week for early passage cells to reach confluence. For most primary human fibroblasts, a density of 2 × 105 cells per 100-mm dish is adequate. Take note of the population doublings of the two samples, using the value of PDtime0 for cells that have just been transduced and then selected.
- Let cells grow for a week at 37°C under 5% CO2.
- Trypsinize, wash, recover in culture media, and then count cells using either a Coulter counter or a hematocytometer.
- Calculate the number of population doublings done by the cells since they were last seeded (or ΔPD), where ΔPD = log[(number of cells recovered) ÷ (number of cell seeded)] / log[2].
- Calculate the current PD of each of the two cell populations by adding the value of their ΔPD to that of their previous PD (at which cells were when they had been plated a week earlier). If ΔPD is negative, set the value of ΔPD to zero.
- Replate cells at the exact same density as in step I and freeze all remaining cells. Label frozen vials with name of strain, vector transduced, population doubling (current PD), and approximate number of cells.
- Repeat steps 2 though 6 until all of the vectortransduced cells have become senescent. For both samples, each cycle of growth will yield a ΔPD that is then added to the previous PD to increase the current PD to its present value.
- For each sample, plot PDn+1 as a function of time. Cells transduced with the empty vector should eventually reach a plateau corresponding to telomerecontrolled senescence, at which point cells will cease to divide. Pursue the experiment until the vectortransduced cells have reached senescence. Cells can be considered senescent once they perform less than a doubling over a 2-week period. Most strains of primary human cells reach senescence after 15-90 doublings. A complete bypass of senescence by the hTERTtransduced cells would then confirm that the cellular life span has been extended by exogenous telomerase. In this eventuality, grow the hTERT-transduced cells until the life span has been extended by a factor of at least 3, at which point the cells can be considered functionally immortal.
The TRAP assay is based on an improved version of the original method described by Kim et al. (1994). It is typically performed using a commercial kit, the TRAPeze telomerase detection kit (Serologicals Corp., Norcross, GA).
Solutions
- 50 mg/ml BSA: Dissolve 0.5 g of BSA in 10 ml of deionized water. Aliquot and store at-20°C.
- 0.5M EDTA: To make 500ml, add 93.1 g of EDTA (disodium salt) to 400ml of deionized water. Adjust pH to 8.0 using sodium hydroxyde. Complete to 500ml with deionized water.
- 6X loading dye: To make 10ml, combine 6.4ml of deionized water, 3 ml of glycerol, 600 µl of 0.5 M EDTA, 25 mg of bromphenol blue, and 25 mg of xylene cyanol.
- 12.5% acrylamide gel solution: To make 50ml, combine 15.6ml of a 40% acrylamide:bisacrylamide [19:1] solution with 5 ml of 5X TBE. Complete to 50 ml with deionized water. Just before casting the gel, add 50µl of TEMED and 500µl of 10% ammonium persulfate. Mix well and cast immediately.
- 5X TBE: To make 1 liter, add 54g of Tris base, 27.5 g of boric acid, and 20ml of 0.5M EDTA, pH 8.0, to 800ml of deionized water. Mix until solute has dissolved. If needed, readjust pH to 8.1-8.5. Complete to 1 liter with deionized water.
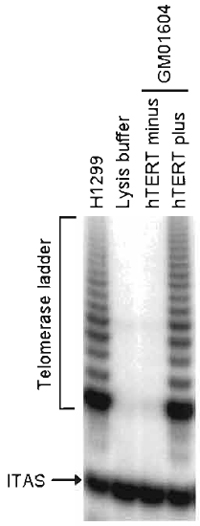 |
| FIGURE 1 Telomerase activity in hTERT-transduced fibroblasts. GM01604 fibroblasts engineered to express exogenous hTERT or no hTERT were assayed for telomerase activity using the TRAPeze telomerase detection kit. H1299 and lysis buffer, respectively, serve as positive and negative controls. ITAS, internal TRAP assay standard. |
- Prepare the following samples for analysis: uninfected cells, hTERT-transduced cells, vectortransduced cells, and a positive control (telomerasepositive cancer cell line, such as 293, H1299, or HeLa cells). Trypsinize and count cells; for each sample, transfer 5 × 104 cells to an Eppendorf tube.
- Pelet cells by low-speed centrifugation (2000g for 5min) and remove supernatant. Spin sample once more for just a few seconds so that the very last drop of remaining medium can be removed with the use of a micropipette. Freeze pellets at -80°C.
- Thaw cell pellets on ice. To each sample, add 100µl of ice-cold TRAPeze kit CHAPS lysis buffer (10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.1 mM benzamidine, 5 mM β-mercaptoethanol, 0.5% CHAPS, 10% glycerol). Disperse cells by forcing the pellets 5-10 times through the tip of a P200.
- Incubate cell lysates on ice for 30min.
- Spin samples in a microcentrifuge at 12,000g for 20min at 4°C.
- Transfer 80µl of the supernatant into a fresh Eppendorf tube. Cell extracts should now contain 500 cell equivalents per microliter. Keep on ice and use within the hour or else freeze and store at -80°C.
- End label the Telomerase Substrate (TS) primer (5'-AATCCGTCGAGCAGAGTT-3'). To prepare a sufficient amount of TS primer for six telomerase reactions, combine 6µl of TS primer, 1.2µl of 10X kinase buffer (provided with the enzyme), 3µl water, 1.5µl of [γ-32P]ATP (3000Ci/mmol, 10mCi/ml), and 0.3µl of T4 polynucleotide kinase (10 units/µ). Incubate at 37°C for 20min. Kill the enzyme at 85°C for 5 min.
- Prepare a master mix containing all components of the telomerase assay, minus the extracts to be tested. To prepare sufficient master mix for six assays, combine 30µl of 10X TRAP reaction buffer (200mM Tris-HCl, pH 8.3, 15mM MgCl2, 630mM KCl, 0.5% Tween-20, 10mM EGTA), 3µl of 50mg/ml BSA, 6µl of dNTP mix (2.5mM each dATP, dTTP, dCTP, and dGTP), 6µl of TRAP primer mix (contains the PCR primers needed for amplifying the telomerase products), 234.6µl of water, 12µl of 32P-labeled TS primer (from the previous step), and 2.4µl of Taq DNA polymerase (5 units/µl). Mix well. Aliquot the master mix in four RNase-free PCR tubes at 49µl/tube.
- On ice, thaw all of the cell extracts to assay. Samples should include the uninfected cells, vectortransduced cells, hTERT-transduced cells, a negative control (TRAPeze kit 1X CHAPS lysis buffer), and a positive control (telomerase-positive cancer cell line).
- Add 1 µl of sample per tube (500 cell equivalents), mix, and place in the thermocycler.
- Incubate at 30°C for 30min.
- Perform the following two-step PCR for 27-30 cycles: 94°C for 30s, followed by 59°C for 30s. While these conditions work on most thermocyclers, optimization of the annealing temperature and addition of a 72°C extension step may be necessary on certain machines.
- Add 10µl of 6X loading dye to all samples. Store at -20°C in a β blocker.
- Prepare a vertical 12.5% polyacrylamide gel. Choose glass plates, spacers, and comb so that the gel will be 1.5mm in thickness and 10-12cm in length. Once the mold is ready, add TEMED and ammonium persulfate to the 12.5% acrylamide gel solution and pour the gel immediately.
- Once the gel has polymerized, load 30 µl of each sample per lane. Run at 400V for 90min or until the xylene cyanol has run 70-75% of the gel length.
- Dispose of the electrophoresis buffer as liquid radioactive waste. Wrap gel in saran wrap.
- Expose gel to an X-ray film or PhosphoImager cassette.
- The presence of an active telomerase should yield a ladder of products with 6-bp increments, starting at 50 nucleotides. For the TRAP assay to be valid, the following conditions should first be met: (1) the negative control corresponding to the lysis buffer should display no such ladder; (2) the positive control (e.g., 293, H1299, or HeLa cells) should yield an intense ladder; and (3) all samples should display a 36-bp band corresponding to the internal TRAP assay standard (ITAS), a control template included in the TRAP primer mix whose amplification serves to document the efficiency the PCR step of the protocol. The lack of ITAS amplification would suggest that inhibitors of Taq DNA polymerase may have been present in some of the samples. If the assay has met these conditions, analysis of the experimental samples can now proceed. A successful reconstitution of telomerase activity by exogenous hTERT should produce a telomerase ladder similar in intensity to that of the positive control, with no such activity detected in either vector-transduced cells or uninfected cells.
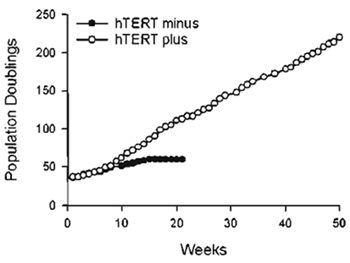 |
| FIGURE 2 Extension of cellular life span by exogenous hTERT. GM01604 fibroblasts engineered to express exogenous hTERT or no hTERT were maintained in continuous log-phase growth and counted once a week. Population doublings executed expressed as a function of time. |
1. Primary Human Fibroblasts
Primary human fibroblasts (GM01604; Coriell Institute, Camden, NJ) were infected with pBabePurohTERT retroviruses under the conditions described in this article. Infected cells were selected for 2 weeks with 750ng/ml puromycin and were then tested for telomerase activity using the TRAPeze telomerase detection kit. Figure 1 shows a strong telomerase ladder in both hTERT-transduced cells and positive control H1299. The 36-bp ITAS is visible in all lanes, indicating that inhibitors of Taq DNA polymerase, which could have resulted in false negatives, were absent. The life span of hTERT-transduced cells was then compared to that of uninfected cells. Cells were maintained in continuous log-phase growth and counted once a week. Figure 2 is a graphic representation of the cumulative number of PD executed by the cells as a function of time. Note that hTERT minus cells ceased dividing at PD 60 whereas hTERT-transduced cells grew beyond PD 180, at which point these cells were considered functionally immortal.
- Other retroviral vectors carrying hTERT are available from Dr. Robert A. Weinberg. (Whitehead Institute for Biomedical Research, Cambridge, MA). These additional vectors include an alternate version of vector pBabePuro-hTERT, as well as plasmid pCIneo-hTERT, a retroviral construct that confers resistance to G418. Be sure to use a vector carrying an hTERT cDNA that has not been epitope tagged at the C terminus, as these modifications can block the access of telomerase to the telomeres (Ouellette et al., 1999).
- To adapt the protocol described for other primary human cells, replace complete medium X with a culture medium that is compatible with the long-term growth and survival of your target cells. Transfected ΦNX-A cells can be made to produce viruses in a large variety of culture media.
- It should be noted that hTERT alone may not be sufficient to immortalize all types of primary human cells. First, the enzyme telomerase does not appear to alter the phenotypic properties, such that postmitotic terminally differentiated cells are unlikely to be rescued by the enzyme. Second, certain primary human cells experience additional forms of senescence that are independent of telomere size (Kiyono et al., 1998). It has been suggested that these extra barriers represent a stress response to inadequate culture conditions, in some cases elicited by the loss of mesenchymal- epithelial interactions (Ramirez et al., 2001; Shay and Wright, 2001). In addition to exogenous hTERT, the immortalization of such primary cells might also require a reoptimization of the culture conditions for long-term growth, the cultivation of the cells over feeder layers, or, alternatively, the use of oncogenes that can block pRB function, such as the SV40 large T antigen, HPV type 16 E7, or adenovirus type 5 EIA.
- When working with amphotropic retroviruses, due caution must be exercised in the production, use, and storage of recombinant viruses. Transfected ΦNXA cells, viral supernatants, and all plasticwares that have been in contact with these reagents should be bleached and treated as biohazard.
- When using 32P, appropriate measures must be taken to ensure that the user remains shielded from radiation and that radioactive by-products and wastes are being contained appropriately. Working areas should also be monitored for radioactive contamination.
- When performing the TRAP assay, precautions should be taken to limit PCR contamination. To limit such contamination, electrophoresis analysis of the samples should be run in a separate area away from the bench where samples are prepared and TRAP reactions performed.
References
Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C. P., Morin, G. B., Harley, C. B., Shay, J. W., Lichtsteiner, S., and Wright, W. E. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349-352.
Condon, J., Yin, S., Mayhew, B., Word, R. A., Wright, W. E., Shay, J. W., and Rainey, W. E. (2002). Telomerase immortalization of human myometrial cells. Biol. Reprod. 67, 506-514.
Jiang, X. R., Jimenez, G., Chang, E., Frolkis, M., Kusler, B., Sage, M., Beeche, M., Bodnar, A. G., Wahl, G. M., Tlsty, T. D., and Chiu, C. P. (1999). Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nature Genet. 21, 111-114.
Kiyono, T., Foster, S. A., Koop, J. I., McDougall, J. K., Galloway, D. A., and Klingelhutz, A. J. (1998). Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396, 84-88.
Morales, C. P., Gandia, K. G., Ramirez, R. D., Wright, W. E., Shay, J. W., and Spechler, S. J. (2003). Characterization of telomerase immortalized normal human oesophageal squamous cells. Gut 52, 327-333.
Morales, C. P., Holt, S. E., Ouellette, M., Kaur, K. J., Yan, Y., Wilson, K. S., White, M. A., Wright, W. E., and Shay, J. W. (1999). Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nature Genet. 21, 115-118.
Ouellette, M. M., Aisner, D. L., Savre-Train, I., Wright, W. E., and Shay, J. W. (1999). Telomerase activity does not always imply telomere maintenance. Biochem. Biophys. Res. Commun. 254, 795-803.
Ouellette, M. M., McDaniel, L. D., Wright, W. E., Shay, J.W., and Schultz, R. A. (2000). The establishment of telomeraseimmortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet. 9, 403-411.
Ramirez, R. D., Morales, C. P., Herbert, B. S., Rohde, J. M., Passons, C., Shay, J. W., and Wright, W. E. (2001). Putative telomereindependent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 15, 398-403.
Shay, J. W., and Wright, W. E. (2001). Aging. When do telomeres matter? Science 291, 839-840.
Yang, J., Chang, E., Cherry, A. M., Bangs, C. D., Oei, Y., Bodnar, A., Bronstein, A., Chiu, C. P., and Herron, G. S. (1999). Human endothelial cell life extension by telomerase expression. J. Biol. Chem. 274, 26141-26148.
Yudoh, K., Matsuno, H., Nakazawa, E, Katayama, R., and Kimura, T. (2001). Reconstituting telomerase activity using the telomerase catalytic subunit prevents the telomere shortening and replicative senescence in human osteoblasts. J. Bone Miner Res. 16, 1453-1464.
Support our developers




