In vivo DNA Replication Labeling
DNA replication in higher eukaryotes takes place in a well-defined spatial and temporal manner. Large numbers of replication sites are simultaneously active, creating characteristic replication patterns during progression through S phase (Nakamura et al., 1986; O'Keefe et al., 1992). Each site is typically active for ~45 min, forming a replication focus of some 100 kb up to several Mb in size, consisting of a cluster of 1-10 simultanously firing replicons (Berezney et al., 2000). Because the higher order chromatin structures revealed by replication foci persist through all stages of the cell cycle, they are referred to as ~l-Mb chromatin domains. Importantly, replication timing reflects important functional chromatin features, as the generich, transcriptionally active euchromatin replicates during the first half of S phase, whereas gene-poor, heterochromatic sequences are later replicating (Cremer and Cremer, 2001).
A transient permeabilization that allows the uptake of macromolecules from the surrounding medium can be achieved by a number of methods (for review, see McNeil, 1989), yet not all of them are useful for in vivo replication labeling. Detergents (e.g., Triton X-100, saponin, or digitonin) permeabilize cells effectively, but have detrimental effects on cell viability, whereas more gentle "noninvasive" methods, such as lipofection and osmotic shift, are not efficient in our experience. More suitable regarding loading efficiency and viability are methods creating slight mechanical damage to the cell membrane in the presence of nucleotides. This can be achieved by shaking a labeling solution with small glass beads over a cell layer or by detaching a cell layer with a cell scraper.
II. MATERIALS AND INSTRUMENTATION
Cy3-dUTP (Amersham Bioscience, Cat. No. PA53022), Cy5-dUTP (Amersham Bioscience, Cat. No PA55022), fluorescein-12-dUTP (Roche Cat. No. 1373242), disposable hypodermic needles (e.g., 0.45mm × 25mm), 15 × 15-mm square coverslips, 60/15-mm tissue culture dishes, phase-contrast microscope, appropriate cell culture medium (with HEPES), paper wipes (e.g., Kimwipes lite precision wipes, Kimberly-Clark), fine forceps, live cell chamber with fitting coverslips (e.g., Bioptechs FCS2).
Solutions
Appropriate cell culture medium (with 25 mM HEPES and supplemented with 10% fetal calf serum and antibiotics); labeling solution: 20µM (Cy3- dUTP) or 50µM (Cy5-dUTP, fluorescein-12-dUTP) in medium.
Steps
- Seed cells on small coverslips (15 × 15 mm) and grow them until they reach near confluence.
- Pick up the coverslip with fine forceps, drain excess medium, dry the bottom side of the coverslip briefly with a wipe, and place it into an empty tissue culture dish. This will prevent the coverslip from sliding during the subsequent scratching procedure.
- Add 8-10 µl of the labeling solution onto the coverslip and distribute it evenly over the cells by gently tilting the dish. Surface tension will prevent the solution from running off the coverslip.
- With the tip of a hypodermic needle, apply parallel scratches into the cell layer. For a high fraction of labeled cells, scratches should be performed a few cell diameters apart from each other and cover the complete coverslip (Fig. 1). For optimal coverage, the procedure can be performed under a low magnification phase-contrast microscope (e.g., 5× objective lens). The procedure should not take longer than a few minutes to avoid drying of the cells.
- Add 5ml prewarmed medium and incubate further. Exchange medium after 30-60min to remove nonincorporated nucleotides.
- To obtain nuclei with segregated labeled and unlabeled chromosome territories, the cells are harvested by trypsinization some hours later and cultivated further for two or more cell cycles prior to live cell observation. The day before live cell observations are carried out, seed cells on coverslips fitting the live cell chamber (Fig. 2).
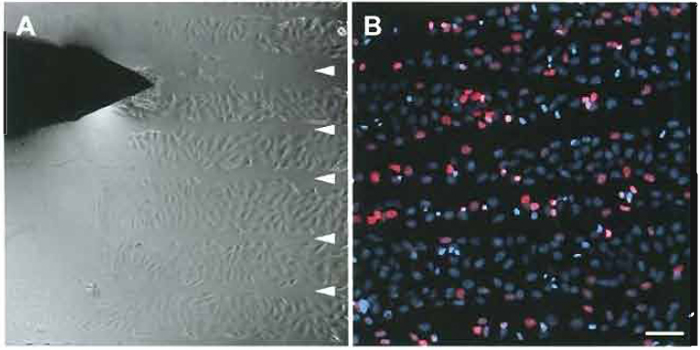 |
| FIGURE 1 (A) Scratch replication labeling of human neuroblastoma cells. Arrowheads indicate scratches in the monolayer applied with the tip of a hypodermic needle (top left) in the presence of Cy3-dUTP. (B) Cells were fixed with 4% formaldehyde two hours after the scratching procedure and counterstained with 4', 6-diamidino-2-phenylindole (DAPI, blue). Numerous cells display Cy3-dUTP labeled nuclei (red) along the scratch lines. Bar: 100µm. |
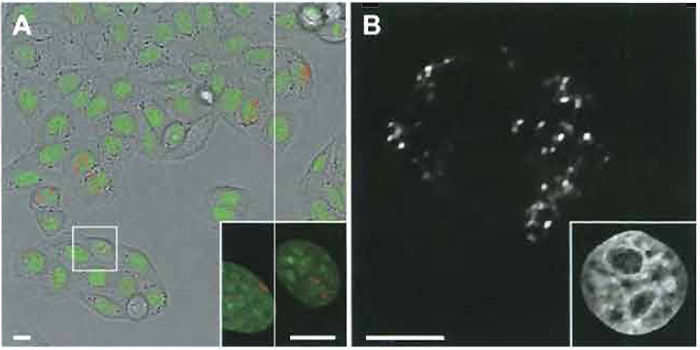 |
| FIGURE 2 (A) Living HeLa cells with green fluorescent protein (GFP)-tagged histone H2B reveal green fluorescent nuclear chromatin. The cell culture was scratch labeled with Cy3-dUTP 5 days prior to observation in the live cell chamber. Numerous cells display nuclei with a few Cy3-1abeled chromosome territories/ ~1-Mb chromatin domains. The framed cells are shown at higher resolution in the inset. Bars: 10µm. (B) Confocal midsection of a HeLa cell expressing histone H2B-GFP fixed 3 days after labeling with Cy3- dUTP shows segregated chromosome territories/~1-Mb chromatin domains at high resolution. (Inset) GFP-labeled chromatin of the same nucleus. Bar: 5 µm. |
The scratch procedure creates damage in the cell membrane that may last for only a few seconds ("transient holes"). This allows the uptake of charged macromolecules (such as fluorochrome-coupled dUTPs) from the surrounding medium, to which cells would normally be impermeable. Accordingly, most S-phase cells damaged alongside the scratch line or lifted off from the surface by the needle will incorporate the modified nucleotides.
The fraction of replication-labeled cells depends on the (1) density of scratches applied, i.e., how many cells are affected, and (2) number of cells that are in S phase. Synchronization of cells at the G1/S transition (e.g., by aphidicolin) is recommended to obtain a high yield of labeled cells.
A pool of fluorescent nucleotides is available for DNA replication over a time scale of roughly l h. The labeling pattern therefore resembles a BrdU-labeling experiment with ≤l-h pulse length.
Cy3-dUTP is significantly brighter and more photostable than Cy5- and fluorescein-dUTP and thus is best suited for in vivo observation. In cases where especially high fluorescence intensities are desirable (e.g., for long-term in vivo observations after segregation), a higher concentration of nucleotides (100 µM) is advantageous. We did not note any improvement of label intensities when adding nonfluorescent dNTPs to the labeling solution or by using different labeling buffers (phosphate-buffered saline or Tris-buffered saline instead of medium).
For studies of nascent RNA formation, the scratch protocol can be applied accordingly using BrUTP (5mM in medium) followed by immunofluorescent detection.
Ansorge, W., and Pepperkok, R. (1988). Performance of an automated system for capillary microinjection into living cells. J. Biochem. Biophys. Methods 16, 283-292.
Berezney, R., Dubey, D. D., and Huberman, J. A. (2000). Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 108, 471-484.
Cremer, T., and Cremer, C. (2001). Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nature Rev. Genet. 2, 292-301.
McNeil, P. L. (1989). Incorporation of macromolecules into living cells. Methods Cell Biol. 29, 153-173.
Nakamura, H., Morita, T., and Sato, C. (1986). Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp. Cell Res. 165, 291-297.
O'Keefe, R. T., Henderson, S. C., and Spector, D. L. (1992). Dynamic organization of DNA replication in mammalian cell nuclei: Spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J. Cell Biol. 116, 1095-1110.
Schermelleh, L., Solovei, I., Zink, D., and Cremer, T. (2001). Twocolor fluorescence labeling of early and mid-to-late replicating chromatin in living cells. Chromosome Res. 9, 77-80.
Walter, J., Schermelleh, L., Cremer, M., Tashiro, S., and Cremer, T. (2003). Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 160, 685-697.
Zink, D., Cremer, T., Saffrich, R., Fischer, R., Trendelenburg, M. E, Ansorge, W., and Stelzer, E. H. (1998). Structure and dynamics of human interphase chromosome territories in vivo. Hum. Genet. 102, 241-251.
Support our developers




