Live-Cell Fluorescent Speckle Microscopy of Actin Cytoskeletal Dynamics & Their Perturbation by Drug Perfusion
Fluorescent speckle microscopy (FSM) is a method used to analyze the movement, assembly, and disassembly dynamics of macromolecular structures in vivo and in vitro (Waterman-Storer et al., 1998). FSM capitalizes on the well-established method of fluorescent analog cytochemistry, in which purified protein is covalently linked to a fluorophore and microinjected or expressed as a green fluorescent protein (GFP) fusion in living cells, incorporated into cellular structures, and whose dynamics are visualized by timelapse or video wide-field epifluorescence microscopy (Wang et al., 1982; Prasher, 1995). This approach has been limited in its ability to report protein dynamics because of inherently high background fluorescence from unincorporated and out-of-focus incorporated fluorescent subunits and the difficulty in detecting movement or turnover of subunits because of their uniform labeling of fluorescent structures. In contrast, FSM offers the capability to measure variations in molecular dynamics in living cells at high spatial and temporal resolution. In addition, FSM reduces outof- focus fluorescence and improves the visibility of fluorescently labeled structures and their dynamics in three-dimensional polymer arrays such as the mitotic spindle (Waterman-Storer and Salmon, 1999; Maddox et al., 2002, 2003).
In its initial development in 1998, FSM utilized conventional wide-field epifluorescence light microscopy and digital imaging with a sensitive, low-noise cooled charge-coupled device (CCD) camera and was applied to study the assembly dynamics and movement of microtubule polymers (Waterman-Storer and Salmon, 1997). Since then, FSM has seen new applications in answering various questions about actin and microtubule cytoskeletal function in vivo during cell motility, neuronal pathfinding, and mitosis, as well as given insight into cytoskeletal dynamics in vitro. It also has been transferred from widefield epifluorescent microscopes to confocal and total internal reflection fluorescence (TIRF) microscopes, which reduce the out-offocus contribution further and thus increase the speckle contrast (Grego et al., 2001; Maddox et al., 2002; Adams et al., 2002; Adams et al., 2004). Computer-based analysis of FSM image series has begun to be developed so that the use of dynamic speckles as quantitative reporters of polymer trajectory, velocity, assembly rate, lifetime, and disassembly rate can be realized (Ponti et al., 2003).
To understand the origin of speckles on microtubules as an example, one must consider how the images of fluorescent microtubules are formed by the optics of the microscope. Microtubules assemble from tubulin dimers into polymers with 1625 dimers per micrometer of microtubule (Desai and Mitchison, 1997). In an image made by a microscope, a fluorescent microtubule appears as wide as the smallest region that the microscope is capable of resolving, defined by geometrical optics of light as r = 0.61λ/NAobj (Inoué and Spring, 1997), where λ is the emission wavelength of the fluorophore and NAobj is the numerical aperture of the microscope objective lens. For example, for Xrhodamine (602nm emission)-labeled microtubules imaged with the best available 1.4 NA optics, the smallest resolvable region of a microtubule is 270nm, which contains 440 tubulin dimers. A low fraction of fluorescent dimers, f = 1%, will produce a mean number n of 4 fluorescent dimers (n = 440 x f) per resolvable image region. In FSM images, the speckle pattern along the microtubule is produced by variations in the number of fluorescent dimers per resolvable image region relative to this mean. Thus, the "contrast" of the speckle pattern can be expressed as the ratio between the standard deviation and the mean of a binomial distribution with n elements:
This formula indicates (i)that the contrast c increases with lowering f and (ii) that it decreases with growing n, indicating the requirement for optics with the highest NA possible.
Thus, time-lapse FSM requires the ability to visualize high-resolution image regions (~0.25µm) containing few (1-10) fluorophores and the capacity to inhibit fluorescence photobleaching. This requires a very sensitive imaging system with little extraneous background fluorescence, very efficient photon collection, a camera with low noise, high quantum efficiency, high dynamic range, high resolution, and suppression of fluorescence photobleaching and photodamage in the specimen with illumination shutters and/or oxygen scavengers (Waterman-Storer et al., 1993, 1998; Mikhailov and Gundersen, 1995).
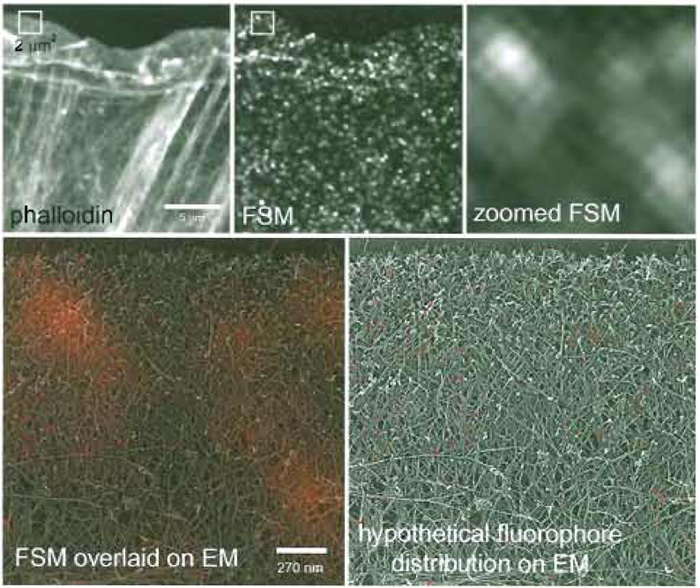 |
| FIGURE 1 FSM of the actin cytoskeleton. (Top) A newt lung epithelial cell was microinjected with a low level of X-rhodamine-labeled actin, fixed, and stained with Alexa-488 phalloidin. In the phalloidin image, all actin filaments are labeled and can be visualized, giving much information about the higher order structural organization, including the location of filament meshworks in the lamellipodium and filament bundles in the cell body. In the single FSM image, much of the structural information is lost, but in time-lapse FSM series, dynamic information is gained that cannot be obtained with higher level labeling of the cytoskeleton. The 2 x 2-µm box in the middle panel is zoomed up in the right-hand panel, and in the bottom right it is colorized red and overlaid onto a quick-freeze deep-etch image of the same-sized region of the actin cytoskeleton in the leading edge of a fibroblast (kindly provided by Tatiana Svitkina, Northwestern University). This gives a sense of the scale of FSM compared to EM so that you can see how many filaments actually fit within the 270-nm resolution-limited image region. In the bottom right panel, a hypothetical fluorophore distribution that could give rise to the speckle pattern is shown, demonstrating the very small proportion of the total actin that is utilized for gaining information in FSM images. |
This chapter article gives guidelines for designing a microscope imaging system for performing time-lapse FSM and protocols for achieving time-lapse FSM imaging of the actin cytoskeleton in living tissue cells cultured on glass coverslips. Finally, it also describes a perfusion system for temporally controlled application of drugs during time-lapse FSM of actin to observe the kinetic evolution of their effects on the dynamics and organization of the actin cytoskeleton.
II. MICROSCOPY INSTRUMENTATION
As opposed to a step-by-step protocol, this section discusses the basic components needed to set up an FSM system, giving the recommendations for critical requirements in each type of component (see Fig. 2).
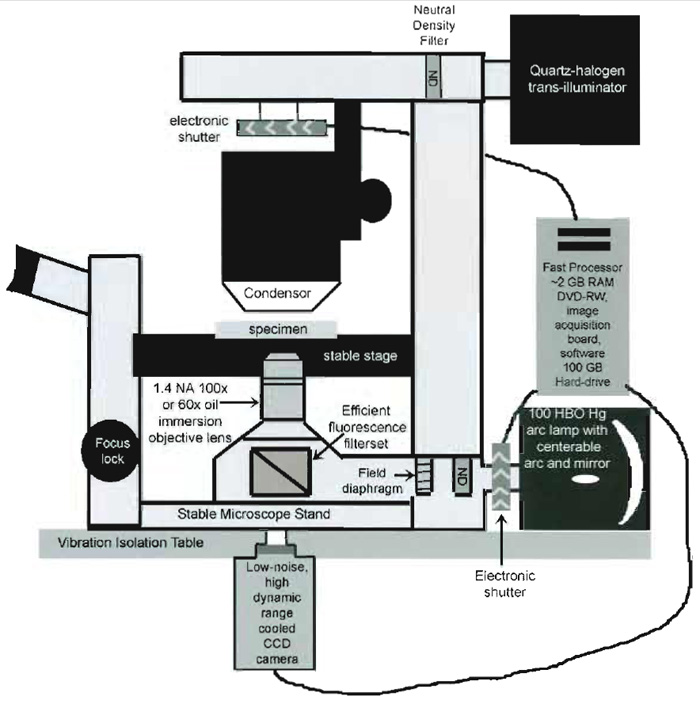 |
| FIGURE 2 Schematic diagram of the important components of an inverted microscope optimized for FSM. |
A. Upright or Inverted Epifluorescent Microscope and Optics
- The microscope stand should be of biological research quality, with a substantial mass that resists vibration. If possible, the microscope should be mounted on a vibration isolation table (available from Newport or TMC). Either upright or inverted configurations can be used, although inverted microscopes offer more flexibility in choosing imaging chambers for living cells and accommodating open petri dishes. The focusing system should be lockable during long-term time-lapse imaging, as slight shifts in focus result in changes in speckle intensity that can be interpreted artifactually as actin dynamics. Acceptable recentmodel uprights include Nikon Eclipse E400, 600, 800, or 1000; Olympus BX series; or Zeiss Axioplan 2. Inverteds include Nikon Eclipse TE-2000 series, Olympus IX series, Zeiss Axiovert 200, or Leica DM IRB.
- The microscope should be equipped with a highquality epi-illuminator. The lamp should be a 100HBO mercury arc, and the lamp housing should include a parabolic mirror and should allow manual control of bulb and mirror centration for proper alignment of Koehler epi-illumination. It is very helpful, although not necessary, to have manual control of centration and size of the field diaphragm of the epi-illuminator. Closing down the field diaphragm to just the area of the specimen being imaged reduces photodamage to the whole specimen and reduces out-of-focus fluorescent flare in the image. Similarly, it is helpful if the epi-illuminator is equipped with slots in which to insert neutral density filters to control specimen illumination and an infrared blocking filter to minimize exposure of the specimen to damaging heat.
- The objective lens (which also acts as the condenser in epi-illumination) should be of the highest numerical aperture available, i.e., 1.4 NA oil immersion. Magnification should be either 60, 63, or 100x, the choice being dependent on the spatial resolution of your camera detector (see Section II,6). Keep in mind that 60 or 63x lenses are often more efficient at transmitting light than 100x lenses. However, you should never sacrifice system resolution for this small gain in light detection. The lens should not contain contrastforming elements such as phase rings that block transmission of photons. The lens should be corrected for chromatic aberration (apochromatic) and flat-field corrected (Plan). We have had good luck using DIC plan apochromat objectives.
- The excitation and emission filters and dichromatic mirror should be as efficient as possible at exciting the fluorophore, separating the excitation from emission, and collecting the emission of the fluorophore of your choice. The use of long-pass instead of band-pass filters may maximize this efficiency. High-quality filter/mirror sets for commonly used fluorophores are available from Chroma Technologies or Omega.
- The path from the objective lens to the detector should be simple and contain as few intervening components as possible to allow maximum photon collection. Removal of analyzers, wave plates, and Wollaston prisms that are used for various modes of polarization microscopy is required. Optovars or magnification changers should optimally be removed; however, if they are needed to match the microscope resolution to the detector resolution (see later), this is a source of light loss that will have to be lived with. The camera port should utilize the most direct path from the specimen. For an upright microscope, this would be directly over the objective, and for an inverted configuration, a bottom port underneath the microscope is the best bet. The bottom port requires a hole in the table upon which the microscope is seated in order to accommodate the camera. If this is impractical, a side camera port, which requires one mirror to direct the image to the camera, is better than a front port that requires at least two mirrors. Finally, prisms that split the image between the ocular and camera port should be removed from the light path.
- The electronically controlled shutter should be mounted with proper adapters in the light path between the lamp house and the epi-illuminator.
- The shutter should be mirrored on the surface facing the lamp to reflect heat away from the specimen.
- The shutter should operate quietly, quickly, reliably, and without excessive vibration. Shutters can be obtained from Vincent Instruments that are actuated from a software-triggered pulse via the serial or parallel port of the computer.
C. Cooled CCD Camera
Choice of the camera is one of the critical, make-orbreak decisions in the design of an FSM imaging system. For imaging the low fluorescence of dim FSM specimens, the camera demands are steep: it should be highly sensitive, extremely low noise (remember that speckles look a lot like noise!), high spatial resolution, high dynamic range, and, depending on your biological application, it may need high speed as well. The camera should be a scientific grade slow-scan cooled chargecoupled device camera. To date, most intensified cooled CCD we have tested (both microchannel plate type or on-chip type) have had noise characteristics that obfuscate speckle detection, although on-nip electron multiplication LCDs appear promising. Cameras are available from several manufacturers (Hamamatsu Photonics, Roper Scientific, Cohu, Andor) at prices ranging from ~$9000 to $30,000 USD. We give an example of the specifications of the camera of choice in our laboratory, the Hamamatsu Orca II ER, as a benchmark.
1. Spatial Resolution
Spatial resolution is determined by the physical size of the silicon photodiodes ("pixels") that convert photons to charge on the CCD chip. These currently range in size from about 6 x 6 to 30 x 30 µm. The larger the pixel size, the more magnification from the microscope will be needed to ensure resolution-limited images. Thus, smaller pixel size is better for FSM, as it will not require photon-robbing magnification changers or optovars in the light path. The total number of pixels making up the CCD and the pixel size will determine the imaging area. For example, our Orca II ER has a 1344 x 1024 array of 6.4 x 6.4 - 41µm2 pixels, resulting in a 8.67 x 6.60-mm CCD capable of imaging an 87 x 66-µm area of the specimen at 100x magnification.
2. Pixel Well Capacity
Based on the physical composition of the silicon photodiodes, pixels will have a maximal number of photoelectrons that it can "hold" before it is saturated with charge, which will correspond to white saturation in the image. The greater this "full well capacity," the greater the potential for a high dynamic range, after taking noise into consideration (see later). The full well capacity is also a function of the pixel size, so for FSM imaging one should consider the best full well capacity per micrometer of pixel area. The Orca II ER has a full well capacity of 18,500 electrons/41 µm2 = 45 electrons per µm2.
CCDs can be illuminated from their front or back sides. Front-side illumination requires that the light pass through substrate materials to reach the photosensitive area, reducing quantum efficiency (QE). This is the configuration of our Orca II ER. Back-illuminated CCDs are physically thinned (also called "backthinned") to allow illumination directly on the photosensitive surface, making them much more sensitive (and much more fragile and expensive!). However, because of the thinning process, there are limits to the size of the pixels, with the smallest currently available at 13 x 13µm. Thus, one has to weigh whether the increased sensitivity is worth the photon loss in having an optavar in the image path, as well as whether one can afford the expense. For FSM applications, sensitive front illuminated CCDs, such as the Orca II ER, work quite well.
4. Spectral Sensitivity
Different types of CCDs have specific probabilities at any given wavelength of converting a photon striking the pixel to an electron that is counted as signal by the camera (quantum efficiency). Manufacturers supply graphs of the wavelength vs QE for their available CCDs. A CCD should be chosen that has a high (>50%) QE in the wavelength range of your fluorophore of choice. The Orca II ER has ~70% QE between 450nm (blue-green) and 600nm (orange-red). Some back-illuminated cameras achieve ~90% QE throughout the visible spectrum, but have excessively large pixels and terribly slow readout (see later).
Once photons are converted to charge in the array of pixels, the charges must be read out to an image acquisition board so that the image can be reconstructed in the computer by assigning a gray value to the relative charge at each pixel position. Charges are transferred out of the CCD in three basic ways. Fullframe readout occurs as each row of pixel charges is transferred serially out of the CCD one row after another. This type is the slowest and introduces the most noise (nonphoton-associated charge) into the image, although can still acceptable for FSM if other camera electronics do not introduce sources of noise. In contrast, in frame transfer and interline transfer CCDs, either the entire pixel charge array or whole rows of pixels are transferred simultaneously to an array of pixels that are masked from light. The charges are then read out from the masked area while the imaging area is being exposed to light again. These types are much faster and less noisy than the fullframe readout type. All three geometries are acceptable for FSM, although attention should be given to the manufacturer's specifications for the noise introduced during readout ("readout noise") as this will deter mine the dynamic range of the camera (see later). Our Orca II ER is the interline transfer type, giving a good balance of higher speed and low readout noise (three to five electrons).
E. Camera Electronics
1. Cooling
Heat on the CCD can cause nonphoton-associated charge to build up in pixels, thus contributing to image noise. The coldest camera possible within a reasonable budget should be chosen. Manufacturers will house the same CCD in cameras with different degrees of cooling, ranging from 20°C below ambient temperature to -50 or-60°C. Do not try to go the inexpensive route here because heat is an avoidable source for noise that can easily mask your very faint FSM signal. The Orca II ER is cooled to -60°C, contributing to its exceptionally low readout noise (see earlier discussion).
In general, the faster the readout speed, the more error is introduced during charge transfer, which translates to noise in your image. Speeds in modern cameras range from 100kHz/pixel in some low-noise back-illuminated cameras to 14-15 MHz/pixel in interline and frame transfer cameras. For quantitative FSM imaging of cytoskeletal dynamics, image acquisition rates of 1-2 images/s may be required, which cannot be accomplished by the slower cameras. Here, a reasonable compromise of speed and low noise must be sought, but it is recommended not to buy a camera much slower than 1MHz. The Orca II ER has a choice of readout speeds, a higher noise, fast readout at 10mHz/pixel or a slower low-noise (quoted earlier) 1.25-mHz/pixel readout rate.
3. Dynamic Range
For FSM imaging of very dim specimens, it is important to have the biggest dynamic range possible (again, within a reasonable budget). Although pixel full-well capacity is set for a given CCD, the number of gray levels this amount of charge is divided up into is not fixed. It can be encoded by 8, 10, 12, 14, or 16 bits of information per pixel, corresponding to 256, 1024, 4096, 16,384, or 65,536 (28 , 210 , 212 , 214 , 216 ) gray levels. However, statistically, it is not possible to distinguish between two gray levels that differ by less than the noise level. Thus the actual dynamic range is determined by the pixel full-well capacity divided by the readout noise. For example, our Orca II ER camera advertises 14 bit dynamic range (16,384 gray levels) and has a full-well capacity of 18,500 photoelectrons. Thus, it must have a noise level of 18,500/16,384 = 1.1 electrons per pixel or less to make use of the full 14 bit range. Because the Orca II ER has a minimum of three electrons noise, the actual dynamic range is 18,500/3 = 6166 discernible gray levels. For FSM imaging, a bigger dynamic range (at least 12 bit) is required so that differences between the intensity of two or three fluorophores can be detected quantitatively.
Being able to read out only a specified portion of the CCD (subarraying) can increase image acquisition speed for imaging small areas of a cell, but it is not necessary. Binning, in which the charges in a group of pixels are combined and read out as a single pixel to increase sensitivity, should not be done in FSM, as this effectively increases pixel size and decreases CCD resolution.
F. Computer, Digital Image Acquisition Board, and Software for Control of Shutter and Image Acquisition
1. Computer
A computer with the fastest processor and most random access memory (RAM) affordable should be used. Currently, computers with ~2-GHz processors and 2-GB RAM can be had for ~$3000 USD. Time-lapse FSM image series are large files often on the order of 500 MB or more and computer "horsepower" is necessary to view and manipulate these. A large hard drive (100 GB) is useful for temporary file storage.
A DVD R/W device is the most economical choice recommended to archive the large files generated by time-lapse FSM. The fastest write speed available within budget should be chosen. A portable USBbased hard drives allow rapid transfer between computers. However, in the best case, a large networked file server is preferred.
3. Image Acquisition Board
Use the board recommended by your camera and software manufacturer, making sure that the board can handle the bit depth of the camera. Many cameras come with their own boards.
4. Software
Software should be capable of time-lapse digital image acquisition and triggering the shutter during camera exposure. The software should allow easy viewing of time-lapse series as movies, with control of play-back rate and adjustment of brightness and contrast in the entire image series. Basic image processing, including the ability to perform low-pass filtering and image arithmetic (subtraction, multiplication, etc.), is required. The software should provide the ability to perform quantitative analysis of intensity, position, and distance. We have used Metamorph (Universal Imaging) with outstanding success. However, NIH-Image freeware (available at http://rsb.info.nih.gov/nih-image/) is also very versatile and many free macros are available.
A critical key to obtaining resolution-limited images of fluorescent speckles is matching microscope and camera resolution. The resolution-limited image region has to be magnified to an area on the CCD large enough to achieve a sampling frequency that is high enough to be able to digitally resolve structures at the resolution limit (Stelzer, 1998). As a rule of thumb, magnifying the resolution limited image region to the size of 3 x 3 pixels on the CCD is sufficient so that the CCD does not limit imaging system resolution or produce aliasing between pixel rows. This is referred to as the Nyquist sampling criterion. Any magnification over this value does not contain significantly more information and simply reduces the area of the specimen that is imaged. The magnification (M) required to achieve this is given by
| M = 3Pwidth/r |
where Pwidth is the width of a pixel and r is the size of the resolution-limited image region. Thus, for red fluorescence with a resolution limit of 0.27µm and a camera with 6.7 µm pixels and a 1.4 NA objective lens, the magnification required to satisfy the Nyquist criterion is 75x. Thus either a 100x objective or a 60x with 1.25x intermediate magnification should be used, whichever transmits more light.
Successful time-lapse FSM imaging of actin dynamics is highly dependent on the choice of cell type. Cells should grow in tissue culture well adhered to glass coverslips and should be relatively large (>50µm diameter), well spread, flat, and thin (<1.0µm). We routinely use PtK1 cells, an epithelial line from ratkangaroo kidney (American Type Culture Collection, Manassas, VA, ccl-35). These cells are optimal as they are large, thin cells and are relatively easy to microinject. PtK1 cells are maintained in F-12 Hams media (Sigma, St. Louis, MO, Cat. No. N-4388) and plated on 22x 22-mm #1.5 cover glasses (Corning, Kennebunk, ME, Cat. No. 2870-22) in 35-mm tissue culture dishes and maintained in a humidified incubator at 37°C supplemented with 5% CO2. Use of this type of coverslips is critical, as high-resolution optics are specifically corrected for this thickness (#1.5 = 0.17mm. thick), and our custom-made perfusion chamber described in Fig. 2 is specifically designed for them. Prior to plating cells, the coverslips are cleaned by sequential washes and sonications in detergent, water, and ethanol, as described in detail in Waterman-Storer (1998). This level of coverslip cleanliness is of paramount importance, as coverslip dirt results in background fluorescence that degrades speckle contrast.
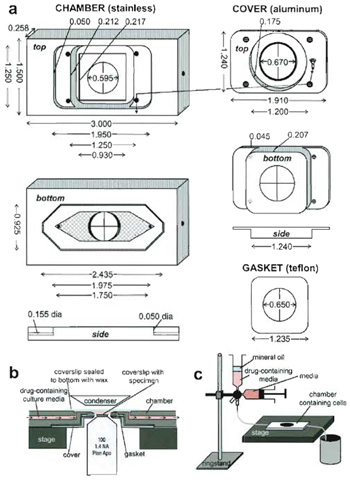 |
| FIGURE 3 A custom-designed perfusion system for drug application during high-resolution FSM imaging. (a) Specifications for chamber design. The chamber is made of stainless steel, the top of aluminum, and the gasket of a 0.005-in. Teflon sheet. Forty-five degree beveled areas are shown in dark gray, holes have crosses across them, and on the bottom of the chamber, the hatched areas are machined-in 0.007 in. to create the imaging chamber. (b) Cutaway side view of the chamber in operation on the microscope stage (not to scale). (c) Schematic of the setup used for drug perfusion. |
For drug treatment studies we have custom designed a live-cell perfusion chamber that allows use of high-resolution oil-immersion objective and condenser lenses and gives near laminar flow for complete exchange of media in a minimal volume while remaining sealed from the environment (Fig. 3). The chamber portion comes in direct contact with media that washes over cells, and thus must be made of inert stainless steel. The chamber top that holds down the coverslip of cells on the chamber has no contact with media, and thus can be made of less inert aluminum, while the coverslip gasket is made of 0.005-in. thick Teflon sheet, for flexibility. A 24 x 60-mm coverslip (Corning, Cat. No. 2940-246) must be sealed to the bottom of the chamber with hot VALAP (a 1 : 1 : 1 mixture of Vaseline, paraffin, and lanolin prepared in the laboratory) to complete the condenser lens-facing side of the imaging chamber. Any good machine shop should be capable of reproducing this chamber based on the diagram in Fig. 3.
IV. PROCEDURES
A. Microinjection of Fluorescent Actin
Microinjection of 1 mg/ml fluorescently labeled actin allows later visualization of protein dynamics by time-lapse FSM.
- Plate cells onto cleaned coverslips (Waterman- Storer, 1998) at least 24h before microinjection.
- Pull microinjection needles from borosilicate glass capillaries (1.0mm outer and 0.78mm inner diameter). Proteins that polymerize, such as actin, are notoriously difficult to microinject, as the large polymers will clog the needle tips. We have ascertained that needles efficient for injecting actin and other polymerizing protein such as tubulin consist of an inner tip diameter of less than 1 µm, and a short, relatively blunt taper to prevent clogging, breaking, or overflexible needles. By experimenting with the parameters of the needle puller, one can adjust the shape of the needle. The following four-step program may serve as a starting point for achieving good needles on the Sutter needle puller, but depending on the age and capabilities of the glass-heating filament in the needle puller, the needle-pulling program will necessitate some variation according to the guidelines in the owner's manual.
Heat Pull Velocity Time Pressure Ramp + 10 100 10 250 500 Ramp 100 10 250 500 Ramp 100 10 250 500 Ramp + 10 100 15 250 500
Commercially available microinjection needles such as Eppendorf Femtotips II work well for actin microinjection and thus are an alternative to pulling your own needles; however, they are expensive and can only be used with Eppendorf injection systems. - In a closed petri dish, rest freshly pulled (or freshly opened commercial) needles on a small cylinder of modeling clay to immobilize them and prevent breaking of the tip. In a chemical fume hood, add a few drops of HMDS to the bottom of the petri dish and close the dish immediately. This silanization vapor on the glass will prevent absorption of protein, therefore reducing needle clogging. Allow at least 2 h incubation of needles in the hood with HMDS before use.
- Just prior to microinjection, chill the microultracentrifuge to 4°C. Thaw an aliquot of labeled actin at body temperature and transfer it to ice immediately. Centrifuge for 20min at 30,000g in a swinging bucket rotor to remove aggregates that may clog the needle.
- Following centrifugation, transfer the tube of labeled actin to ice. Using a Hamilton syringe, back load approximately 1µl of clarified fluorescent actin (being careful not to touch the syringe needle to the pellet!) into a microinjection needle inserted as close as possible to the microneedle tip. Insert the microneedle into the needle holder of the microinjection device. The needle holder should be at an angle of approximately 45° relative to the microscope stage. The back pressure should be set to ~0.8psi. To rid the needle of air bubbles, use the clean button (which transiently increases the back pressure to a maximum of 65 psi) to push solution through the needle tip.
- Center the needle over the objective lens as well as possible by eye and lower the needle into the medium in the 35-mm tissue culture dish, stopping immediately when the needle comes in contact with the medium. Close the condensor diaphragm down and move the needle in the x-y plane so that the very tip of the needle is in the centered cone of light. The needle should be well above the cells, so open the condensor diaphragm and look through the microscope, moving the needle slowly in the xy direction and looking for the out-of-focus shadow of the needle in the image field of view. Once the shadow of the needle is in the field, focus up until the needle is in focus. Now slowly alternate between moving the focus down and then the needle down until the needle is in focus just above the cells. Alternatively, using a Bertrand lens, view the objective lens back aperture, where the tip of the needle will be in focus when the needle has just entered the medium. Center the needle tip in the image of the phase ring, then view the image, and lower the needle until it is in focus. Quickly getting needles in focus at the plane of the specimen without breaking their tips will likely take some practice.
- We find it most simple to keep constant slow flow of labeled actin from the needle tip to help prevent clogging. Bring the flowing needle tip over the cell that you want to inject and lower the needle until it touches the cell just adjacent to the nucleus. As the solution flows into the cell, you will see a change in the contrast of the cell around the nucleus. Rapidly remove the needle from the cell before it blows up.
- After microinjecting approximately 40 cells in a dish successfully, replace the medium with fresh medium, and place the dish back into the incubator for at least 2h to allow the cells to recover from injection and to incorporate fluorescently labeled actin into the cytoskeleton.
- After microinjection of cells, begin preparing for live-cell imaging and drug perfusion. Turn on the heater to preequilibrate stage and objective lens temperatures several hours before you plan on imaging the cells. This will minimize focus drift caused by temperature shifts that expand/contract the metal in the microscope body. Prepare 20ml of media (buffered with 10mM HEPES if necessary) containing 30µl of Oxyrase/ml of media.
- Right before mounting cells in the perfusion chamber, prepare the perfusion setup (see Fig. 3c). Our custom-designed perfusion setup uses two 10-ml syringes mounted into a two-way luer-lock syringe stopcock (available from medical supply houses) with a 25-gauge needle at the bottom, feeding into a tube that connects to the perfusion chamber (Fig. 3). One syringe should be filled with 10ml of drug-free media containing 30 µl of Oxyrase/ml of media. Be sure to tap out all bubbles from the syringe to maintain Oxyrase activity. This syringe should be mounted on the stopcock valve parallel to the tabletop. The second syringe will be used for drug-containing media. Remove the plunger from this syringe and place into the stopcock valve perpendicular to the first syringe (perpendicular to the tabletop). Fill it with 8 ml of drug-treated media supplemented with oxyrase and then cover with mineral oil to protect the oxyrase from the air. The drug perfusion will be done by gravity, as opposed to pushing a syringe.
- To prepare the perfusion chamber, melt VALAP and heat the perfusion chamber on a heating block for a few minutes. Using a cotton-tipped applicator swab, place a thin layer of VALAP around the outer ring on the bottom of the perfusion chamber and adhere a 24 x 60-mm coverslip onto this. Allow the valap to solidify under the cover glass and place on a cool surface to return the perfusion chamber to room temperature.
- Once the perfusion chamber is at room temperature, place vacuum grease around the perimeter of the coverslip-mounting surface of the top side of the chamber. Place the chamber on the microscope stage to allow it to heat to 37°C. Once the perfusion chamber is warm, place the coverslip with injected cells face down onto the chamber. Put the gasket on top of the coverslip, making sure it seals the chamber; an additional layer of vacuum grease may aid in this. Then place the aluminum chamber cover over the gasket and attach it to the chamber by screwing down opposite corners in turns, being careful not to crack the coverslip.
- Once the perfusion chamber is set up, insert the drug-free media inlet tube into one end of the perfusion chamber and hold the chamber so the inlet end is facing down so that when the chamber is filled with media, all air bubbles will be pushed up through the upper outlet. Slowly push the drug-free media through the perfusion chamber. Avoid trapping bubbles by lightly tapping the chamber on a hard surface as you fill it.
- Insert another tube into the outlet end of the perfusion chamber for discarded media to flow into a waste beaker.
- Back the microscope focus all the way down and then place a small drop of the appropriate immersion oil on the 100x 1.4 NA objective lens front element. Place the filled perfusion chamber cell side down on the microscope stage and bring the focus up until the oil on the objective is in contact with the coverslip.
- Bring the cells into focus using phase-contrast or DIC optics, being careful not to smash the objective lens into the coverslip. If using DIC, remember to remove the Wollaston prism and analyzer from the imaging path after finding the cells! Search the coverslip using epifluorescence for the injected cells and verify their viability by seeing if fluorescent actin is excluded from the nucleus. Be careful to keep your cell-gazing time to the absolute minimum, as you will be photobleaching the fluorescent actin. Choose a cell to image that is very dimly fluorescent. Getting the cells in focus with a high-magnification oil-immersed lens and finding the injected cells will take practice and patience.
- Begin FSM image acquisition. Using the microscope system described in Section II with a mercury arc bulb with <200h of use on it, exposure times for FSM images taken with unattenuated (neutral density filters removed) light of 500-1000ms should provide a signal of around 0.3-0.6% of the total gray scale. This translates to a signal of 50-100 gray levels above background for a 14 bit camera or 10-20 for a 12 bit camera. We find that maximizing the gain setting of the camera (if available) is very helpful when taking images of actin speckles. For imaging processes such as actin flow in a PtK1 cell, an acquisition rate of one image every 5-10s will allow accurate actin flow velocity analysis. Microscope control software such as Metamorph can be set up easily to acquire images with specific camera parameters and light exposure times at regular intervals for a prescribed total length of time. Plan on taking predrug images for 5-10 min and postdrug images for several tens of minutes.
- After an appropriate accumulation of images of actin dynamics in the untreated cell, switch the stopcock position to allow drug-containing media to flow through the chamber at a rate of ~2-4ml/min. Drug perfusion should take approximately 30s. Continue to acquire images before, during, and after drug perfusion.
- Continue imaging until the process of interest has terminated or the fluorescent actin is photobleached.
Interpretation of actin speckle dynamics provides information about actin turnover and actin flow in a cell and how this relates to cell behavior. Using image analysis software such as Metamorph, images can be processed and analyzed.
- After acquiring a time series of actin FSM images for at least 5 to 10min, observe the actin movement by simply watching the time series as a movie. This can be done using Metamorph, Quicktime (Apple, Cupertino, CA), or NIH Image. Pay careful attention to the trajectories of actin movement, convergence of actin in different regions of the cell, and speeds of actin movement in different regions of the cell.
- Use kymograph analysis to obtain information about the rate of actin retrograde flow and/or the activity of the leading edge. Kymographs are time-distance plots created by extracting the same row of pixels from each image in a time series and laying them side by side in a montage (see Figs. 4 & 5). For measuring actin dynamics, choose a row of pixels along the trajectory of actin movement as determined from watching the movie. For measuring leading edge activity, choose a row of pixels perpendicular to the leading edge.
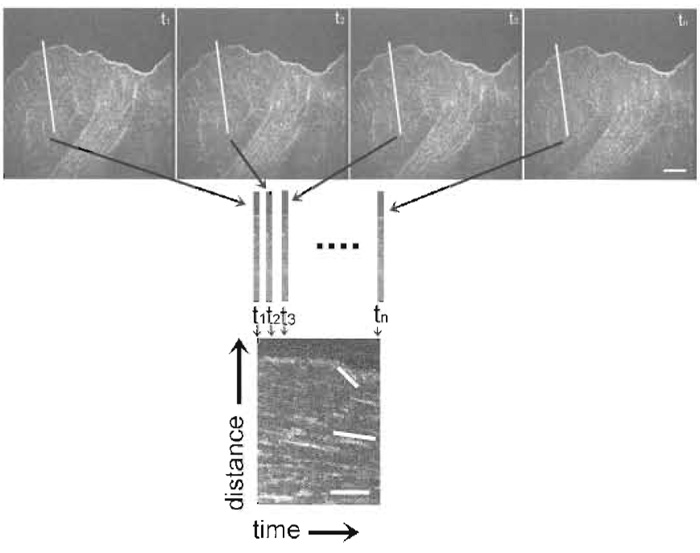 |
| FIGURE 4 Kymograph construction from an FSM time series. The first row of images is taken from a FSM time series. The white line in each image depicts the row of pixels along the trajectory of actin movement (as determined from watching the series as a movie) extracted to construct the kymograph to show actin dynamics, seen below the images. Rows of pixels are laid side by side in a montage to create a time vs distance plot, seen on the bottom of the figure. This kymograph allows analysis of the rates of actin movement within different regions of the cell. Lines in the kymograph on the bottom highlight discreet rates of actin flow in which the magnitude of the slope of the lines relates to the velocity of actin movement. Bar: 10 µm. |
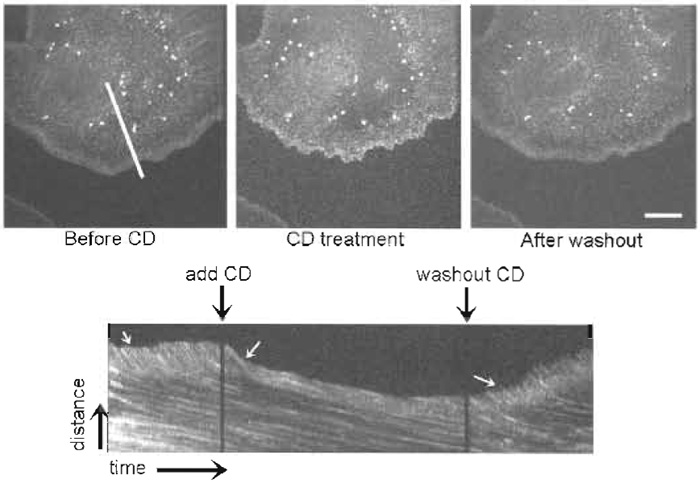 |
| FIGURE 5 FSM reveals that cytochalasin D (CD) halts actin polymerization and fast retrograde flow in the lamellipodium. Images are taken from a time series before CD treatment, during CD treatment, and after washing out CD. (Bottom) A kymograph taken from the time series along the white line indicated in the first image. Perfusion and washing out of CD are marked by black arrows over the kymograph. White arrows on the kymograph indicate the region of rapid retrograde flow inhibited by CD treatment and relieved after CD washout. Bar: 15 µm. |
Successful FSM imaging of live cells requires several critical components, such as proper fluorescent protein concentration, successful suppression of photobleaching, and high-quality, well-labeled, fully functional fluorescent protein. Fluorescent actin speckles cannot be detected if the concentration of fluorescent protein in the cell is too high, which can occur if the microneedle concentration is >1mg/ml. Even at proper needle concentrations, too much fluorescent actin can get into the cell if the microinjection technique is not optimized. Conversely, if there is not enough fluorescent protein injected into cells, the camera exposures required to acquire a decent image will be excessively long and may result in motion artifacts within the image. Photobleaching is also a major pitfall in FSM imaging. This can occur if the imaging chamber is not fully sealed, if there are air bubbles present in the chamber, or if the Oxyrase has begun to lose its potency. Ensuring an airtight chamber with fresh Oxyrase will reduce photobleaching greatly. Finally, ensuring that the actin remains functional during the labeling process is an extremely important consideration for quality FSM imaging. If you are making your own fluorescent actin, be sure to use high-quality acetone powder that is finely powdered or "fluffy" and that the dye is fresh and has been stored in dessicant at -20°C. Old dye will render actin nonpolymerizable, so avoid using dye that is not fresh.
References
Adams, M. C., Matov, A., Yarar, D., Gupton, S. L., Danuser, G., and Waterman-Storer, C. M. (2004). Siginal analysis of total internal reflection fluorescent speckle microscopy (TIR-FSM) and widefield epi-fluorescence FSM of the actin cytoskeleton and focal adhesions in living cells. J Microsc. 216, 138-52.
Adams, M. C., Salmon, W. C., Gupton, S. L., Cohan, C. S., Wittmann, T., Prigozhina, N., and Waterman-Storer, C. M. (2003). A highspeed multispectral spinning-disk confocal microscope system for fluorescent speckle microscopy of living cells. Methods. 29(1), 29-41.
Bulinski, J. C., Odde, D. J., Howell, B. J., Salmon, T. D., and Waterman-Storer, C. M. (2001). Rapid dynamics of the microtubule binding of ensconsin in vivo. J. Cell Sci. 114(Pt 21), 3885-3897.
Desai, A., and Mitchison, T. J. (1997). Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 13, 83-117.
Gupton, S. L., Salmon, W. C., and Waterman-Storer, C. M. (2002). Converging populations of f-actin promote breakage of associated microtubules to spatially regulate microtubule turnover in migrating cells. Curr. Biol. 12(22), 1891-1899.
Inoué, S., and Spring, K. R. (1997). "Video Microscopy." Plenum Press, New York.
Maddox, P., Desai, A., Oegema, K., Mitchison, T. J., and Salmon, E. D. (2002). Poleward microtubule flux is a major component of spindle dynamics and anaphase in mitotic Drosophila embryos. Curr. Biol. 12(19), 1670-1674.
Maddox, P., Straight, A., Coughlin, P., Mitchison, T. J., and Salmon, E. D. (2003). Direct observation of microtubule dynamics at kinetochores in Xenopus extract spindles: Implications for spindle mechanics. J. Cell Biol. 162(3), 377-382.
Maddox, P. S., Moree, B., Canman, J. C., and Salmon, E. D. (2003). Spinning disk confocal microscope system for rapid highresolution, multimode, fluorescence speckle microscopy and green fluorescent protein imaging in living cells. Methods Enzymol. 360, 597-617.
Mikhailov, A. V., and Gundersen, G. G. (1995). Centripetal transport of microtubules in motile cells. Cell Motil. Cytoskel. 32(3), 173-186.
Ponti, A., Vallotton, P., Salmon, W. C., Waterman-Storer, C. M., and Danuser, G. (2003). Computational analysis of f-actin turnover in cortical actin meshworks using fluorescent speckle microscopy. Biophys. J. 84(5), 3336-3352.
Prasher, D. C. (1995). Using GFP to see the light. Trends Genet. 11(8), 320-323.
Small, J. V. (1981). Organization of actin in the leading edge of cultured cells: Influence of osmium tetroxide and dehydration on the ultrastructure of actin meshworks. J. Cell Biol. 91(3 Pt 1), 695-705.
Stelzer, E. H. K. (1998). Contrast, resolution, pixelation, dynamic range and signal-to-noise ratio: Fundamental limits to resolution in fluorescence light microscopy. J. Microsc. 189, 15-24.
Svitkina, T. M., Verkhovsky, A. B., McQuade, K. M., and Borisy, G. G. (1997). Analysis of the actin-myosin II system in fish epidermal keratocytes: Mechanism of cell body translocation. J. Cell Biol. 139(2), 397-415.
Verkhovsky, A. B., Svitkina, T. M., and Borisy, G. G. (1999). Selfpolarization and directional motility of cytoplasm. Curr. Biol. 9(1), 11-20.
Watanabe, N., and Mitchison, T. J. (2002). Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science 295(5557), 1083-1086.
Waterman-Storer, C. M. (2002a). Fluorescent speckle microscopy (FSM) of microtubules and actin in living cells. In "Current Protocols in Cell Biology" (K. S. Morgan, ed.) Wiley, New York.
Waterman-Storer, C. M. (2002b). Microtubule/organelle motility assays. In "Current Protocols in Cell Biology" (J. S. Bonifacio, M. Dasso, J. B. Harford, J. Lippincott-Schwartz, and K. M. Yamada, eds.). Wiley, New York.
Waterman-Storer, C. M., Desai, A., Bulinski, J. C., and Salmon, E. D. (1998). Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr. Biol. 8(22), 1227-1230.
Waterman-Storer, C. M., and Salmon, E. D. (1997). Actomyosinbased retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J. Cell Biol. 139(2), 417-434.
Waterman-Storer, C. M., and Salmon, E. D. (1998). How microtubules get fluorescent speckles. Biophys. J. 75(4), 2059-2069.
Waterman-Storer, C. M., and Salmon, E. D. (1999). Fluorescent speckle microscopy of microtubules: How low can you go? FASEB J. 2, S225-S230.
Waterman-Storer, C. M., Salmon, W. C., and Salmon, E. D. (2000). Feedback interactions between cell-cell adherens junctions and cytoskeletal dynamics in newt lung epithelial cells. Mol. Biol. Cell 11(7), 2471-2483.
Waterman-Storer, C. M., Sanger, J. W., and Sanger, J. M. (1993). Dynamics of organelles in the mitotic spindles of living cells: Membrane and microtubule interactions. Cell Motil. Cytoskel. 26(1), 19-39.
Support our developers




