Syringe Loading: A Method for Assessing Plasma Membrane Function as a Reflection of Mechanically Induced Cell Loading
In the past, we have described a method for loading large macromolecules into the cytoplasm of cultured cells via the production of transient plasma membrane wounds inflicted by defined amounts of fluid shear stress (Clarke and McNeil, 1992, 1994). This technique is referred to as "syringe loading" and has been utilized to load a variety of macromolecules (i.e., fluorescent dextrans, proteins, immunoglobulins, calcium indicator dyes, plasmid DNA, and antisense oligonucleotides) into various different cell types (i.e., endothelial cells, fibroblasts, epithelial cells, lymphocytes, and amoeba). This technique is very simple and straightforward, relying on the capacity of the plasma membrane to reseal after the infliction of a transient plasma membrane disruption. During the syringe loading procedure, the mechanical force applied to the cells to produce plasma membrane wounding is fluid shear stress generated as a consequence of the cell suspension being forced through a narrow orifice in the form of a 30-gauge hypodermic needle. The macromolecule to be loaded is dissolved in the suspension medium and enters the cell cytoplasm across a diffusion gradient during the time the plasma membrane wound is open. As such, this technique for macromolecular loading of cells in suspension is simple, reproducible, and inexpensive.
Although complex in nature, the membrane wound response in toto can be quantified using direct end point measures that describe the final outcome of the process. Such measures include cell survival at a given level of mechanical perturbation, the number of wounded cells present in the surviving cell population, the amount of membrane wound marker that enters the wounded cell, and the relative size of the membrane wound created based on membrane wound marker size. If membrane wounds are produced in a defined and reproducible manner, the effects of various environmental conditions on plasma membrane function, as reflected by alterations in the membrane wound response, can be quantified by the end point measures described earlier. We have used this approach to probe the effects of different gravitational conditions on the biophysical properties of the plasma membrane in cultured adherent cells using a technique known as impact mediated loading (Clarke et al., 2001). This article describes the use of syringe loading as a means of investigating the effects of environmental conditions on the plasma membrane wound response in suspension cells.
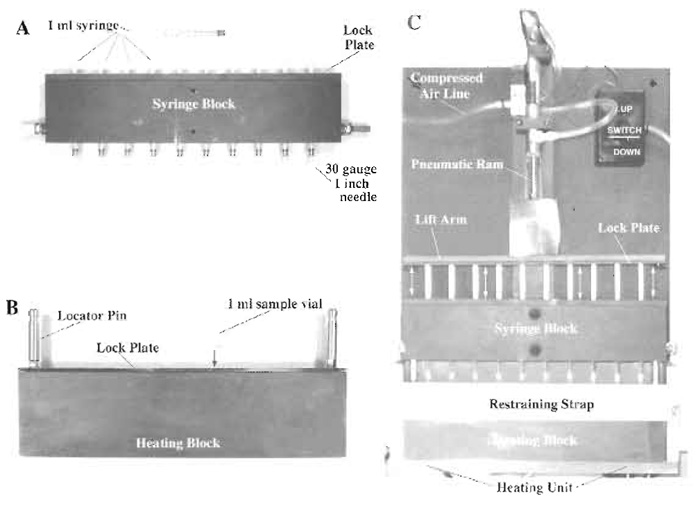 |
| FIGURE 1 Multisample syringe loader. Ten, sterile, 1-ml disposable syringes are secured in the syringe block using a lock plate. A sterile, 30-gauge, 1-in. hypodermic needle is affixed to the tip of each syringe, making sure that the needle is seated firmly (PlA). The syringe block is then attached to the pneumatic ram assembly of the main body of the device with the syringe plungers being secured directly to the lift arm using a lock plate (Pl C). Ten 1-ml sample vials, loaded with 0.5 ml of cells suspended in FDx loading solution, are loaded into a heating block (preequilibrated to 37°C) and secured in place using a lock plate (Pl B). The heating block containing the sample vials is then returned to its heating unit (set at 37°C) in the main body of the device and is secured in place with its restraining strap (Pl C). The pneumatic ram is placed in the "down" position so that the syringes are closed, and the whole syringe block is lowered and locked in place via the locator pins present on the top of the heating block. This arrangement ensures accurate registering of the each hypodermic needle into the center of its corresponding sample vial. The cell suspension in loading solution is pulled up into the syringe barrel by activating the pneumatic ram in the "up" direction and is expelled from the syringe by activating the pneumatic ram in the "down" direction. Expulsion pressure is controlled by a pressure valve attached to the compressed air line that feeds the ram assembly (Pl C). |
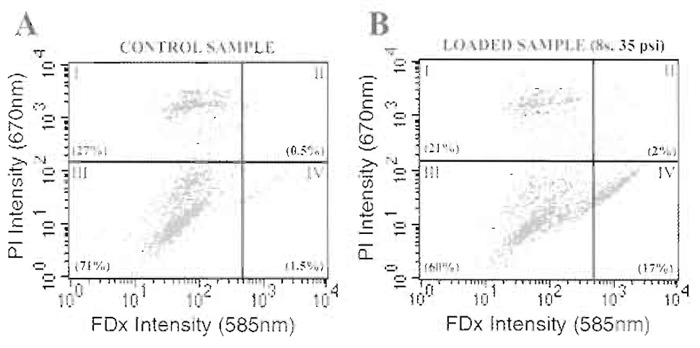 |
| FIGURE 2 Dual-channel fluorescent flow cytometry analysis for the simultaneous determination of membrane wounding and irreparable membrane damage in suspension cells. Control samples, consisting of cell suspensions processed in an identical fashion to experimental samples other than they are not syringe loaded, are used to determine quadrant analysis parameters. Dead or dying cells, present even in the control, nonsyringe loaded sample, stain positively for PI (i.e., quadrants I and II) (PlA). After syringe loading at 35 psi expulsion for a total of eight strokes (Pl B), loaded cells stain positively for FDx (i.e., quadrants II and IV). Truly wounded cells (i.e., FDx positive and PI negative), which have completely resealed their plasma membrane disruptions before the addition of PI to the suspension, are positive only for FDx (i.e., quadrant IV). Numbers in parentheses are values (%) for the cells falling in each quadrant. |
Dulbecco's modified Eagle's medium (X1 concentration) (DMEM, Cat. No. 320-1885AG), bovine calf serum (CS) (Cat. No. 200-6170AG), and penicillin-streptomycin solution (Cat. No. 600- 5140AG) are obtained ready to use from Gibco BRL (Grand Island, NY). Fluorescein isothocyanate-labeled dextran (Mr10 kDa) (FDx) (Cat. No. D-1821) and propidium iodide (Cat. No. P21493) are obtained from Molecular Probes (Eugene, OR). Tissue culture flasks (T75 and T25) (Cat. Nos. 10-126-41 and 10-126-26) and sterile polypropylene conical centrifuge tubes (50-ml capacity) (Cat. No. 05-538-55A) are obtained from Fisher Scientific (Pittsburgh, PA). Sterile polypropylene conical sample vials (1-ml capacity) (Cat. No. 05- 538-55A) are obtained from National Scientific Company (Quakertown, PA). Disposable 1-ml syringes (Cat. No. 9602) and 30-gauge hypodermic needles (Cat. No. 305128) are from Becton-Dickinson (Rutherford, NJ). The multisample syringe loader described in Fig. 1 was built by a local machine shop to specifications provided by the authors. As such, sample vial dimensions can be varied depending on experiment requirements, including the use of sterile, septum-sealed sample vials.
III. PROCEDURES
A. Preparation of Tissue-Cultured Cells for Syringe Loading
Stock Solutions and Media Preparation
- Stock FDx solution: Add 200mg of dry FDx powder to 1 ml of serum-free DMEM and vortex periodically over a period of 30min to achieve complete solubilization. Centrifuge this solution at 10,000g for 10min at room temperature to remove any undissolved FDx and sterilize, if desired, by ultrafiltration. Stock FDx (200mg/ml) can be stored at 4°C in the dark for up to a month or aliquoted and frozen in the dark at -80°C for storage up to a year.
- Stock propidium iodide solution: Add 100 mg of dry PI powder to 1 ml of serum-free DMEM and vortex periodically over a period of 30min to achieve complete solubilization. Centrifuge this solution at 10,000 g for 10 min at room temperature to remove any undissolved PI and collect the supernatant as a stock solution. Stock PI (100mg/ml) can be stored at 4°C in the dark for up to a month or aliquoted and frozen in the dark at -80°C for storage up to a year.
- FDx loading solution: Add 250µl of stock FDx solution to 4.75 ml of serum-free DMEM to obtain final concentrations of 10mg/ml FDx. Use the solution immediately.
- 5% CS.DMEM: Add 5 ml of sterile penicillin/streptomycin solution and 50ml of sterile CS to 445ml of sterile (X1) DMEM solution to obtain DMEM culture medium containing 5% CS, 100IU/ml penicillin, and 100µg/ml streptomycin (5% CS.DMEM). Store at 4°C for up to 21 days.
- Grow Jurkat cells to confluence in T75 (75cm2) culture flasks using 15 ml of 5 % CS.DMEM maintained at 37°C in a 5% CO2 humidified atmosphere with subculture every third day. Carry out subculture by removing the cell suspension from the T75 flask and placing it in a sterile 50-ml centrifuge tube followed by centrifugation at 100g (~550rpm) for 5min at room temperature. Decant the spent medium, resuspend the cell pellet gently in 30ml of fresh 5% CS.DMEM, and dispense equal volumes into two fresh T75 flasks.
- On the day of the experiment, collect cells by centrifugation as in step 1. Gently resuspend cells in 10 ml of 5% CS.DMEM and determine cell number using a hemacytometer or electronic cell counting device (i.e., a Coulter particle counter) and adjust the cell density to 2 × 106 cells/ml. Aliquot 5 ml of this cell suspension into T25 flasks in preparation for radiation exposure.
- Expose cells to different doses of gamma irradiation using a Gammacell 1000 (Cs137 source). Our experiments were performed at Baylor College of Medicine (Houston, Texas).
- Incubate cells at 37°C in a 5% CO2 humidified atmosphere for a period of 2h.
- Remove cells from T25 flasks and collect each sample by centrifugation in sterile 50-ml centrifuge tubes at 100g (~550rpm) for 5min at room temperature.
- Carefully remove medium from the cells and filter through a 0.2-µm cellulose acetate filter to remove any remaining cell debris from the supernatant. Aliquot the medium in 1-ml aliquots and store at -80°C in the dark for subsequent biochemical testing, such as determination of lipid peroxidation marker production.
- Resuspend each cell pellet in 2ml of warm serum-free DMEM and determine the cell number by counting in a hemacytometer/Coulter counter.
- Adjust the cell density of the cell suspension to 1 × 106 cells/ml by the addition of an appropriate amount of warm serum-free DMEM.
- Dispense 0.5-ml samples of this cell suspension into a minimum of 13 sterile conical polypropylene 1-ml sample vials.
- Add 25µl of prewarmed stock FDx solution to all sample vials, mix by gentle vortexing, and load 10 of the 13 sample vials into the heating block (prewarmed to 37°C) of the multisample syringe loader (Fig. 1B).
- Immediately perform syringe loading on the samples from step 10 while the remaining three vials are incubated at 37°C in ambient air as control samples for FDx uptake by pinocytosis and cell loss due to processing.
B. Syringe Loading Protocol
- Place heating block containing 10 sample vials containing identical aliquots of the cell suspension into the multisample syringe loader heating unit and attach the restraint strap as shown (Fig. 1C).
- Insert the 10 separate 1-ml syringes with attached 1-in.-long, 30-gauge needles (which have been loaded previously into the syringe block, Fig. 1A) into the vials and lock in place using the guide posts (Fig. 1C).
- Draw the cell suspension up into the barrel of the sterile syringes through the 30-gauge hypodermic needles by activating the pneumatic ram attached to the syringe plungers using the "up" switch (Fig. 1C). Set the pneumatic ram so that the syringe plungers do not move any further up than the 0.5-ml mark on the syringe barrels.
- Once the barrels of the syringes are filled (which takes approximately 2s), expel the cell suspensions through the 30-gauge needles back into their respective sample vials at a constant pressure of 35psi by reversing the direction of the pneumatic ram using the "down" switch. This procedure is defined as two strokes.
- Repeat this procedure three more times so that each cell suspension has been subjected to eight strokes under identical expulsion pressure conditions.
- Remove the test sample vials from the multisample syringe loader.
- Take all the samples (including the control samples, which have been incubated in FDx loading solution at 37°C but not syringe loaded), add 0.5 ml of 5% CS.DMEM to each vial, collect the cells by centrifugation at 100g for 5 min at room temperature, and remove the supernatant (i.e., FDx loading solution).
- Add 0.5ml of warm 5% CS.DMEM to each cell pellet and resuspend the cells by gently vortexing the sample and store at 37°C in a 5% CO2 humidified atmosphere in preparation for analysis. Analysis should be started as soon as possible.
- Immediately prior to fluorescent flow cytometry analysis, add a 10-µl aliquot of stock PI solution (final PI concentration of 1 mg/ml) to each sample and mix by gentle vortexing. Add PI to each individual sample immediately before analysis.
- Analyze control samples first by dual-channel fluorescent flow cytometry using a Becton-Dickinson FACSCalibur system (or similar fluorescent flow cytometer) and plot a two-axis scatter plot of cell fluorescent intensity at 585nm (FDx signal) and 670nm (PI signal) using the associated software. These control samples are used to define "quadrant" gating thresholds for the control cell population with regard to FDx and PI background signals (Fig. 2). Collect a total of 10,000 events for analysis of each sample.
- Analyze syringe loaded samples by dual-channel fluorescent flow cytometry using the quadrant gates defined earlier (Fig. 2). Collect a total of 10,000 events for analysis of each sample.
- Calculate the number of dead or dying cells in each sample (expressed as a percentage of the total cells analyzed, Fig. 3A) by counting the number of cells in the sample that are positive for PI staining, including those cells that are also positive for FDx (i.e., Fig. 2, quadrants I and II, respectively).
- Calculate the number of wounded cells (expressed as a percentage of the total cells analyzed, Fig. 3B) and the mean fluorescent value (MFV; Fig. 3C) of the wounded population by determining the number and staining intensity of cells that are positive for FDx staining only (i.e., Fig. 2, quadrant IV).
- Calculate a wound index (which reflects cell survival of mechanically induced plasma membrane damage, the number, and the MFV of the wounded cells) as described previously (Clarke and McNeil, 1992) for each radiation dose (Fig. 3D), where the wound index = % dead cells × % wounded cells × mean fluorecent value / 1000.
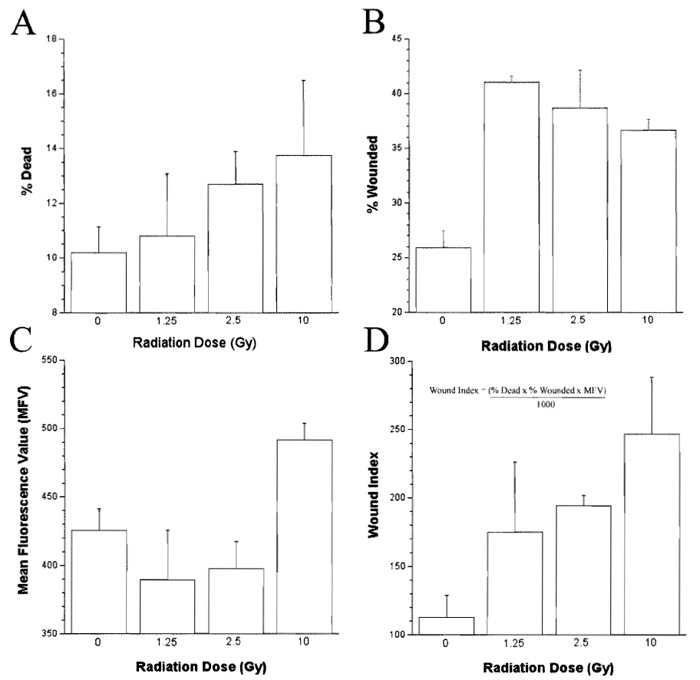 |
| FIGURE 3 Dose-dependent effects of gamma irradiation on the syringe loading-induced membrane wounding response of the lymphoblastic cell line Jurkat. |
The technology described herein allows the effects of a wide range of environmental conditions and their potential countermeasures (e.g., radiation exposure and radio-protectants) to be assessed in a variety of cell suspension models, including peripheral blood lymphocytes from human subjects. A less refined version of this approach has been used previously to study the effects of increasing membrane cholesterol content on plasma membrane susceptibility to mechanical shear-induced membrane wounding (Clarke et al., 1995a). The apparatus and protocol described earlier have taken what was essentially a macromolecular loading technique and adapted it for use as a means of assessing the effects of radiation exposure on the membrane function of lymphoblastic cells in suspension. The technique also has the advantage of being able to immediately discriminate between dead and dying cells and truly wounded cells in suspension after the application of mechanical shear force using two-channel fluorescent flow cytometry (Fig. 2).
The apparent dose response in membrane wound susceptibility observed relative to radiation exposure in our model indicates that gamma irradiation induces alterations in the plasma membrane components of Jurkat cells, if not immediately, then most certainly within 2h of exposure (Fig. 3). This time course of events suggests that these effects are not associated with genomic damage. The effects of radiation exposure on membrane wounding observed in this study are similar to that described previously after an increase in plasma membrane order. One possible reason for such an increase in membrane order after radiation exposure is the production of reactive oxygen species (ROS), such as superoxide, hydrogen peroxide, hydroxyl, peroxyl, and alkoxyl radicals, which may lead to membrane damage and consequent cross-linking of membrane components (Clarke et al., 2003). This concept is supported by the experimental observation that Jurkat cells exposed to gamma irradiation not only exhibit an increase in susceptibility to membrane wounding within 2h, but also produce significant amounts of lipid peroxidation markers (data not shown).
- Jurkat cells are relatively fragile in tissue culture. Cells should be handled gently with a minimum of agitation, being especially careful when performing manipulations that require pipetting or vortexing. This fragility is evidenced by the relatively large number of PI-positive cells (i.e., dead or dying cells) present in control samples that have not been subjected to syringe loading (see Fig. 2A).
- Jurkat cells should be used for syringe loading experiments 1 day after subculture to ensure the optimal number of healthy cells in the culture.
- When calculating the theoretical shear stress imposed during syringe loading, it is important to determine the flow rate through the needle experimentally. This will change primarily depending on the expulsion pressure and the number of cells in the suspension; both parameters are under the control of the investigator. These wounding parameters need to be optimized for each particular cell type.
- It is important to prevent excessive pH changes caused by exposure of the bicarbonate-buffered culture medium to atmospheric air during the procedure as these are potentially harmful to the cells.
Clarke, C. H., Weinberger, S. R., and Clarke, M. S. F. (2003). Application of ProteinChip array technology for detection of protein biomarkers of oxidative stress. Crit. Rev. Oxid. Stress Aging 1, 366-379.
Clarke, M. S., Bamman, M. M., and Feeback, D. L. (1998). Bed rest decreases mechanically induced myofiber wounding and consequent wound-mediated FGF release. J. Appl. Physiol 85(2), 593-600.
Clarke, M. S., Caldwell, R. W., Chiao, H., Miyake, K., and McNeil, P. L. (1995a). Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ. Res. 76(6), 927-934.
Clarke, M. S., and McNeil, P. L. (1992). Syringe loading introduces macromolecules into living mammalian cell cytosol. J. Cell Sci. 102(Pt. 3), 533-541.
Clarke, M. S. E, and McNeil, P. U (1994). Syringe loading: a method for inserting macromolecules into cells in suspension. In "Cell Biology: A Laboratory Handbook" (J. E. Celis, ed.), pp. 30-36, Academic Press, San Diego.
Clarke, M. S. F., Pritchard, K. A., Medows, M. S., and McNeil, P. L. (1995b). An atherogenic level of native LDL increases endothelial cell vulnerability to shear-induced plasma membrane wounding and consequent FGF release. Endothelium 4, 127-139.
Gimpl, G., Burger, K., and Fahrenholz, F. (1997). Cholesterol as modulator of receptor function. Biochemistry 36(36), 10959-10974.
Kuroda, Y., Ogawa, M., Nasu, H., Terashima, M., Kasahara, M., Kiyama, Y., Wakita, M., Fujiwara, Y., Fujii, N., and Nakagawa, T. (1996). Locations of local anesthetic dibucaine in model membranes and the interaction between dibucaine and a Na+ channel inactivation gate peptide as studied by 2H- and 1H-NMR spectroscopies. Biophys. J. 71(3), 1191-1207.
McNeil, P., and Terasaki, M. (2001). Coping with the inevitable: How cells repair a torn surface membrane. Nature Cell Biol. 3, E124-E129.
McNeil, P. L., and Steinhardt, R. A. (1997). Loss, restoration, and maintenance of plasma membrane integrity. J. Cell Biol. 137(1), 1-4.
Miyake, K., and McNeil, P. L. (1995). Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J. Cell Biol. 131(6 Pt 2), 1737-1745.
Needham, D., and Nunn, R. S. (1990). Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J. 58, 997-1009.
Pritchard, K. A., Schwarz, S. M., Medow, M. S. and Stemerman, M. B. (1991). Effect of low-density lipoprotein on endothelial cell membrane fluidity and mononuclear cell attachment. Am. J. Physiol. 260, C43-C49.
Reidy, M. A., and Lindner, V. (1991). Basic FGF and growth of arterial cells. Ann. N. Y. Acad. Sci. 638, 290-299.
Tanii, H., Huang, J., Ohyashiki, T., and Hashimoto, K. (1994). Physical-chemical-activity relationship of organic solvents: Effects on Na(+)-K(+)-ATPase activity and membrane fluidity in mouse synaptosomes. Neurotoxicol. Teratol. 16(6), 575-582.
Whiting, K. P., Restall, C. J., and Brain, P. E (2000). Steroid hormoneinduced effects on membrane fluidity and their potential roles in non-genomic mechanisms. Life Sci. 67(7), 743-757.
Yu, Q. C., and McNeil, P. L. (1992). Transient disruptions of endothelial cells of the rat aorta. Am. J. Pathol. 141, 1349-1360.
Zhelev, D. V., and Needham, D. (1993). Tension-stabilized pores in giant vesicles: Determination of pore size and pore line tension. Biochim. Biophys. Acta 1147(1), 89-104.
Support our developers




