Use of Brain Cytosolic Extracts for Studying Actin-Based Motility of Listeria monocytogenes
Calcium chloride (Cat. No. 102378), magnesium chloride hexahydrate (Cat. No. 105832), potassium chloride (Cat. No. 4936), HEPES (Cat. No. 10110), sucrose (Cat. No. 1.07654), sodium chloride (Cat. No. 1.06404), disodium hydrogen phosphate dihydrate (Cat. No. 1.06580), sodium dihydrogen phosphate hydrate (Cat. No. 6346), and paraffin (Cat. No. 1.07160) are from Merck. EGTA (Cat. No. E-3889), methylcellulose (Cat. No. M-0555), ATP (Cat. No. A-2383), erythromycin (Cat. No. E-6376), and lanoline (Cat. No. L-7387) are from Sigma. Chymostatin (Cat. No. 17158), leupeptin (Cat. No. 51867), pepstatin (Cat. No. 52682), PEFABLOCK (Cat. No. 31682), aprotinin (Cat. No. 13178), and creatine kinase (Cat. No. 127566) are from Boehringer-Ingelheim. Dithiothreitol (DTT, Cat. No. 43815), Tris-HCL (Cat. No. 93363), and glycerol (Cat. No. 49770) are from Fluka. Cell-Tak (Cat. No. 354241) is from BD Biosciences. Creatine phosphate (Cat. No. 621714) is from Roche. Vaseline (Cat. No. 16415) is from Riedel-de Haen. Brain heart infusion (BHI) culture medium and Bacto agar are from Difco Laboratories. Rhodamine-labelled actin (Cat. No. AR05) is from Cytoskeleton. A glass homogeniser equipped with a Teflon piston, glass slides (76 × 26 mm), glass coverslips (22 × 22mm), forceps, scissors, and razor blades are from local suppliers. Bacterial motility is observed using an Axiovert 135TV microscope (Zeiss) equipped with a Plan-Apochromat 100×/1.4 NA oil immersion objective. Images can be acquired with a cooled, back-illuminated CCD camera (TE/CCD-1000 TKB; Roper Scientific) driven by IPLab Spectrum software (Scanalitics).
A. Explantation of Mouse Brains
Solution
Phosphate-buffered saline (PBS): 130mM NaCl, 1.5mM NaH2PO4·H2O, and 4mM Na2HPO4·2H2O, pH 7.4. For 1 litre, weigh out 7.65g NaCl, 0.21g NaH2PO4·H2O, and 0.72 g Na2HPO4·2H2O. Dissolve in 900 ml H2O, adjust pH to 7.4 with 1N NaOH, and bring volume to 1 litre. Store at 4°C.
Steps
- Kill mice using CO2 or by cervical dislocation.
- Remove skin and fur from the head using thintipped scissors and discard.
- Cut the skull bone by sliding the scissors along the sagittal suture (Fig. 1a, dashed red line).
- Cut the frontal, parietal, and interparietal bones along their sutures and remove them (Fig. 1a, dashed green line).
- Gently pinch the surface of the brain to lift the meninges up and gently ease the brain out of the skull.
- Put explanted brains in ice-cold PBS.
- Wash brains three times in ice-cold PBS to remove tissue debris and blood residues.
- Proceed to Section III,B or flash freeze the brains in liquid nitrogen. Store frozen brains at -80°C.
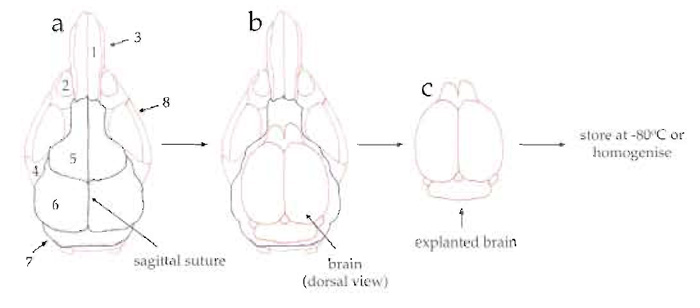 |
| FIGURE 1 Diagram of the explantation of mouse brains. (a) Dorsal view of mouse skull showing the nasal bone (1), eye socket (2), nasal process of incisive bone (3), zygomatic process (4), frontal bone (5), parietal bone (6), interparietal bone (7), and zygomatic bone (8). (b) Dorsal view of skull after cutting (along the dashed green and red lines) and removing the frontal, parietal, and interparietal bones. (c) Dorsal view of explanted brain. |
Solutions
- Homogenisation buffer (HB): 20mM HEPES, pH 7.5, 100mm KCl, 1 mM MgCl2·H2O, 1 mM EGTA, and 0.2mM CaCl2. For 1 litre, weigh out 4.76g HEPES, 7.45g KCl, 0.2g MgCl2·H2O, 0.38g EGTA, and 0.02g MgCl2. Dissolve in 900ml H2O, adjust pH to 7.5 with 1N NaOH, and bring volume to 1 litre. Store at 4°C.
- Protease inhibitors: 20mg/ml chymostatin, ling/ ml leupeptin, 1mg/ml pepstatin, 167mM Pefabloc, and 10mg/m! aprotinin. To prepare stock solutions, dissolve 1mg chymostatin in 50 µl dimethyl sulfoxide, 0.5mg leupeptin in 500µl H2O, 0.5mg pepstatin in 500µl methanol, 20mg Pefabloc in 500µl H2O, and 0.5mg aprotinin in 50µl H2O. Aliquot and store at -20°C.
- 0.1M ATP: For 50ml, weigh out 2.75g ATP. Dissolve in H2O, aliquot, and store at -20°C.
- 0.1M DTT: For 50ml, weigh out 0.77g DTT. Dissolve in H2O, aliquot, and store at -20°C.
- 2M sucrose: For 20ml, weigh out 13.69 g sucrose. Dissolve in H2O, aliquot, and store at -20°C.
- Homogenisation buffer supplemented with protease inhibitors (HBI): HB containing 60µg/ml chymostatin, 5 µg/ml leupeptin, 10 µg/ml pepstatin, 4mm Pefabloc, 2µg/ml aprotinin, 0.5mM ATP, and 1mm DTT. Shortly before use add to 20ml of HB 60µl of 20mg/ml chymostatin, 100µl of 1mg/ml leupeptin, 200µl of 1mg/ml pepstatin, 476µl of 167mM Pefabloc, 4µl of 10mg/ml aprotinin, 200µl of 0.1M DTT, and 50µl of 0.1M ATP.
- After the last wash in PBS (see Section III,A, step 7) remove PBS and weigh the brains.
- Cut the brains into small pieces using a razor blade (keep brains on ice).
- Add 0.75ml of HBI per gram of wet tissue (keep brain suspension on ice).
- Transfer brain suspension into a glass homogeniser on ice.
- Grind brain tissue for 20 passages of the pestle on ice.
- Centrifuge crude extract at 15,000g for 1h at 4°C.
- Recover clarified supernatant (cytosolic brain extract) and supplement it with 150mM sucrose, 50mg/ml creatine kinase, 30mM creatine phosphate, and 0.5 mM ATP.
- Aliquot and flash freeze in liquid nitrogen. Store frozen aliquots at -80°C.
C. Preparation of Bacteria
Solutions
- Brain heart infusion (BHI) broth: Prepare liquid medium according to the manufacturer's instruction, autoclave, filter, and store at 4°C. For agar plates, add Bacto-agar (15g/liter of BHI broth), autoclave, and pour 30ml in a 10-cm petri dish. Store plates at 4°C.
- Erythromycin stock solution: Dissolve 50mg of erythromycin in 10ml of pure ethanol. Store at 4°C.
- Streak the bacteria onto BHI agar plates. Incubate at 37°C for 24h.
- Put 5ml of BHI (supplemented with 50µg/ml erythromycin) in a 15-ml sterile Falcon tube. Scrape a few colonies off the BHI plate using a sterile pipette tip or a flamed bacteriological loop. Inoculate the broth and grow bacteria overnight at 37°C with vigorous shaking.
- Transfer bacterial culture to a centrifuge tube and pellet the bacteria at 10,000g for 3 min.
- Wash bacterial pellet three times in homogenisation buffer. After the final washing step, resuspend pellet in a final volume of homogenisation buffer corresponding to the initial volume of bacterial culture.
- Alternatively, supplement the overnight culture with 20% glycerol, aliquot, and store at -80°C.
D. Listeria Motility Assay
- 2% methycellulose in homogenisation buffer (stock solution): Heat 100ml of HB to 60°C and then add 2g of methylcellulose. Stir vigorously until the methycellulose dissolves. Cool down and store at room temperature.
- 0.5% methycellulose in homogenisation buffer (working solution): To make 10 ml, mix 7.5 ml of HB with 2.5 ml of 2% methycellulose. Store at 4°C.
- VALAP: Mix vaseline, lanoline, and paraffin in a 1:1:1 ratio (w/w/w) and homogenise at 75°C. Store at room temperature.
- G buffer: 5mM Tris-HCl, pH 7.6, 0.5mM ATP, 0.1 mM CaCl2, and 0.5 mM DTT. For 1 litre, weigh out 0.8 g Tris-HCl, 0.01 g CaCl2, and then add 5 µl of 0.1M ATP and 5µl of 0.1M DTT. Dissolve in 900ml H2O, adjust pH to 7.6 with 1N NaOH, and bring volume to 1 litre. Store at 4°C.
- Rhodamine-labelled actin: Add 6µl of G buffer to one aliquot of rhodamine-labelled actin. Mix gently and store on ice.
Steps
- Wash bacteria three times in homogenisation buffer. Resuspend pellet in 20µl homogenisation buffer.
- In a small Eppendorf tube, mix 4 µl brain extract, 4 µl of 0.5% methycellulose, 0.5 µl bacteria suspension, and 0.2µl rhodamine-labelled actin. Mix by pipetting up and down gently. Do not vortex. Incubate mixture at room temperature for 10min.
- Remove 1.7 µl of motility mixture and spot it onto a glass slide. Gently place a 22 x 22-mm glass coverslip over the drop and press down until the drop spreads to the edges. Seal the coverslip edges with VALAP.
- Observe slide with an upright or inverted microscope. Actin comet tails can be observed easily by phase contrast or epifluorescence using a Plan- Apochromat 100x/1.4NA oil immersion objective (Figs 2 and 3).
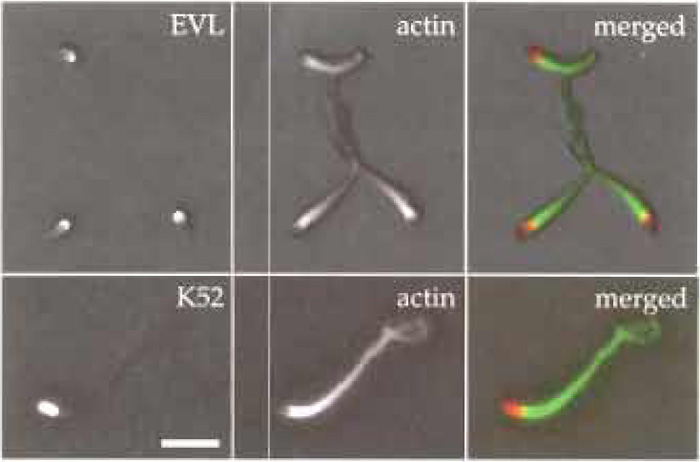 |
| FIGURE 2 Immunofluorescence microscopy showing Listeria actin tails formed in mouse brain extracts. Bacteria were incubated in mouse brain extract for 30min at room temperature and then 5µl of motility mixture was applied onto Cell Tak-coated cover slips (the coating procedure was done according to the manufacturer's instructions) and incubated for 5min on ice. Afterwards, bacteria were fixed with 4% PFA for 20min at room temperature. Immunolabelling was done according to Geese et al. (2002) using the affinity-purified polyclonal antibody K52 to label bacterial surface, the monoclonal antibody 84H1 to label EVL, and Texas redlabelled phalloidin to label the actin tails. Primary antibodies were detected using Alexa 488-conjugated secondary antibodies. Scale bar: 5 µm. |
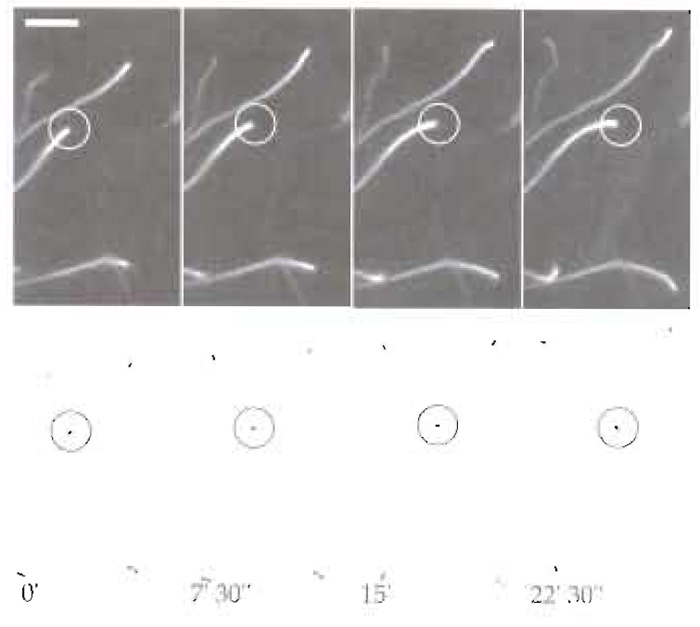 |
| FIGURE 3 Dynamics of Listeria motility in mouse brain extracts. Bacteria were mixed with 4 µl brain extract, 4 µl of 0.5% methycellulose, and 0.2 µl rhodamine-labelled actin and were incubated at room temperature for 10min. Thereafter, 1.7µl of motility mixture was spotted onto a glass slide and a 22 × 22-mm glass coverslip overlaid on it. Bacterial motility was observed using a Axiovert 135 TV inverted microscopy equipped with phase-contrast and epifluorescence optics using a Plan-Apochromat 100×/1.4NA oil immersion objective. Images were acquired using a cooled, back-illuminated CCD camera (TE/CCD-1000 TKB; Roper Scientific) driven by IPLab Spectrum software (Scanalitics). (Top) Rhodamine-labelled actin and (bottom) the corresponding phase-contrast images. Scale bar: 10µm. |
Although cell-free systems based on egg extracts of Xenopus laevis or human platelet extracts provide excellent in vitro systems for supporting actin-based bacterial motility, their ability to do so can be affected negatively by various factors, such as health of eggs or quality and age of platelet preparations. In this context, mouse brain extracts offer a more "robust" in vitro system that does not seem to be influenced by external factors such as mouse strain and age.
The reconstitution of Listeria motility using mouse brain extracts can have a variety of uses. For instance, it can be used to study the role of actin cytoskeletal components involved in Listeria motility by interfering with their function using specific inhibitors or antibodies, as described in May et al. (1999). Moreover, the procedure described here may be further developed and adapted to obtain cell-free extracts from normal cultured cells or cells that lack or express mutated versions of the actin cytoskeletal proteins of interest, thus widening the spectrum of in vitro systems available for studying bacterial motility.
Acknowledgments
I thank David A. Monner and Jürgen Wehland (GBF, Department of Cell Biology) for helpful discussions and valuable support.
References
Cossart, E, and Bierne, H. (2001). The use of host cell machinery in the pathogenesis of Listeria monocytogenes. Curr. Opin. Immunol. 13, 96-103.
Frischknecht, F., and Way, M. (2001). Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 11, 30-38.
Laurent, V., and Carlier, M. F. (1998). Use of platelet extracts for actinbased motility of Listeria monocytogenes. In "Cell Biology: A Laboratory Handbook" (J. Celis, ed.), pp. 359-365. Academic Press, New York.
Laurent, V., Loisel, T. P., Harbeck, B., Wehman, A., Grobe, L., Jockusch, B. M., Wehland, J., Gertler, F. B., and Carlier, M. F. (1999). Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J. Ceil Biol. 144, 1245-1258.
Lingnau, A., Chakraborty, T., Niebuhr, K., Domann, E., and Wehland, J. (1996). Identification and purification of novel internalin- related proteins in Listeria monocytogenes and Listeria ivanovii. Infect. Immun. 64, 1002-1006.
Loisel, T. P., Boujemaa, R., Pantaloni, D., and Carlier, M. F. (1999). Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature 401, 613-616.
Marchand, J. B., Moreau, P., Paoletti, A., Cossart, P., Carlier, M. F., and Pantaloni, D. (1995). Actin-based movement of Listeria monocytogenes: Actin assembly results from the local maintenance of uncapped filament barbed ends at the bacterium surface. J. Cell Biol. 130, 331-343.
Pistor, S., Chakraborty, T., Walter, U., and Wehland, J. (1995). The bacterial actin nucleator protein ActA of Listeria monocytogenes contains multiple binding sites for host microfilament proteins. Curr. Biol. 5, 517-525.
Pistor, S., Grobe, L., Sechi, A. S., Domann, E., Gerstel, B., Machesky, L.M., Chakraborty, T., and Wehland, J. (2000). Mutations of arginine residues within the 146-KKRRK-150 motif of the ActA protein of Listeria monocytogenes abolish intracellular motility by interfering with the recruitment of the Arp2/3 complex. J. Cell Sci. 113, 3277-3287.
Skoble, J., Auerbuch, V., Goley, E. D., Welch, M. D., and Portnoy, D. A. (2001). Pivotal role of VASP in Arp2/3 complex-mediated actin nucleation, actin branch-formation, and Listeria monocytogenes motility. J. Cell Biol. 155, 89-100.
Small, J. V., Stradal, T., Vignal, E., and Rottner, K. (2002). The lamellipodium: Where motility begins. Trends Cell Biol. 12, 112-120.
Smith, G. A., Theriot, J. A., and Portnoy, D. A. (1996). The tandem repeat domain in the Listeria monocytogenes ActA protein controls the rate of actin-based motility, the percentage of moving bacteria, and the localization of vasodilator-stimulated phosphoprotein and profilin. J. Cell Biol. 135, 647-660.
Theriot, J. A., Rosenblatt, J., Portnoy, D. A., Goldschmidt-Clermont, P. J., and Mitchison, T. J. (1994). Involvement of profilin in the actin-based motility of L. monocytogenes in cells and in cell-free extracts. Cell 76, 505-517.
Support our developers




