Video Enhanced Contrast Microscopy
Video contrast enhancement is a technique that considerably improves images obtained by a large variety of video-based imaging systems used in light, electron, and confocal microscopy. It requires a camera system with manually adjustable "gain" and "offset" (also called "pedestal" or "black level" by some manufacturers). Much better results are obtained if digital image processing can be applied in addition. In light microscopy the most striking image improvements are possible if the technique is applied to images obtained by polarized light methods. In such cases the improvement in true resolution is almost 2-fold and in visualization is 10-fold over conventional light microscopy by eye. The specimens most suitable for observation by this technique are unstained, low-contrast objects, such as living cells, uncontrasted semithin electronmicroscopic sections, or gels and colloidal material. The objects made additionally accessible to observation include organelles, especially small vesicles, isolated microtubules (25nm), latex beads 20nm in size and larger, and colloidal gold particles of 5 nm in diameter and larger. The basic principles and theory are described in detail elsewhere (Weiss et al., 1999; Weiss and Maile, 1993).
Video-enhanced contrast (VEC) microscopy requires the combination of a versatile, high-quality research microscope with an analog and digital realtime image processor. The parts required were discussed in detail elsewhere (Inoué and Spring, 1997; Weiss et al., 1999; Weiss and Maile, 1993) so that a short summary may suffice here.
To utilize the technique to its full extent, a massive, stable microscope preferably equipped with differential interference contrast (DIC), and if required additional epi-illumination fluorescence, transmitted light bright-field, dark-field, or other techniques, is used. The illumination should be as bright and even as possible to provide enough light for working at the highest magnifications. A stabilized DC mercury arc or xenon lamp with a narrow band filter (e.g., green 546 ± 10nm for mercury arc lamps) is recommended; with some microscopes it may preferably be equipped with a light-scrambling fiber optic connection (Ellis, 1985; Inoué, 1986) (Technical Video Ltd.). The condenser and objective should be of the oil immersion type when working with objective magnifications of 40x and more. The numerical aperture (NA) of the condenser front lens must match that of the objective; for 60 and 100x objectives, a 1.4 NA oil condenser front lens is required. An exit port projecting 100% of the light to the video camera is essential for fluorescence and high magnification DIC work. In order to visualize objects of sizes near and below the limit of resolution, optical magnification of 2 or 4x is in addition to 100 or 60X, NA 1.3-1.4 objectives required and is obtained by the use of high oculars (16, 25x) and a video camera objective (50 or 63mm) or, preferably, by direct projection of the image to the camera and additional magnification changers. This yields magnifications on a monitor with a 25-cm screen width of 10,000x and more. A temperature-controlled box is needed for work with cultured cells. The use of at least one heat-absorbing and one heat-reflecting filter is essential; a narrow band green filter and a UV filter are recommended for DIC work with most cell types.
The image is picked up by a high-quality video camera, usually a chalnicon or newvicon tube camera. Charge-coupled device (CCD) cameras are equally suitable only if they have a comparable dynamic range. The cameras need to have a manually adjustable offset (black level) and gain to allow for analog contrast enhancement.
III. STRATEGY OF IMAGE GENERATION
Video contrast enhancement of microscopic images obtained using bright-field, dark-field, anaxial illumination including Varel contrast, fluorescence, reflected light confocal, or fluorescence confocal optics, rather than DIC and polarization optics, is very straightforward. It is performed by following the steps in Table I with the exception of
step 4.
 |
Most striking image improvements are usually gained with DIC and polarization microscopy, but special attention and some explanations are required for step 4. Allen et al. (1981a,b) and Inoué (1981, 1989; Inoué and Spring, 1997) simultaneously developed procedures of video contrast enhancement for polarized- light techniques that differed considerably in their approach but yielded very similar results. Although this is not the place to judge which one is more appropriate, we have to distinguish clearly between the two strategies to avoid confusion.
Allen named his techniques "Allen video-enhanced contrast" differential interference contrast and polarization (AVEC-DIC and AVEC-POL, respectively) microscopy. AVEC techniques involve the introduction of additional bias retardation by setting the polarizer and analyzer relatively far away from extinction to gain a high specimen signal. Allen suggested the use of a de Sénarmont compensator (Bennett, 1950), which consists of a quarter-wave plate (specific for the wavelength used) in front of a rotatable analyzer. In DIC microscopy, alternatively but less accurately, the desired bias retardation can also be introduced by shifting the adjustable Wollaston prism. Allen recommended a bias retardation of one-quarter to one-ninth of a wavelength away from extinction, with one-ninth (20°) as the best compromise between high signal and minimal diffraction anomaly of the Airy pattern (Allen et al., 1981b). The enormous amount of stray light introduced at such settings is removed by an appropriately large setting of analog and/or digital offset.
Thus in IVEC microscopy, stray light is not admitted, as the polarizers stay close to extinction and the special rectifying optics further reduce the stray light. Consequently, filters to reduce brightness are not required. On the contrary, a very bright arc lamp, ideally with a fiber-optic illuminator, is necessary to saturate the camera. The AVEC technique, however, electronically improves primary optical images characterized by low contrast and relatively high stray light content, arising from a "non-optimal" optical arrangement. In IVEC microscopy, no compromise is made regarding the optics, and consequently less demanding electronic steps are required to rescue the image. The AVEC technique is, however, one that can be used with any good research microscope equipped with commercial film polarizers.
Generally, it is most desirable in video microscopy to obtain near-saturation of the video camera prior to applying analog or digital enhancement. If there is insufficient light, the following measures are recommended: use brighter lamps (mercury or xenon lamp); redo the illumination adjustments such as setting Köhler illumination and centering the filament or arc, possibly while observing the image on the monitor to improve; remove ground glass diffusers from the light path; make sure that the video exit port receives 100% of the light; reduce magnification slightly; or replace the otherwise highly recommended narrow band-pass filter by a wider one.
In the case of excessive light reaching the camera, the following steps are recommended: apply weak neutral density gray filters, employ high-power light sources that can be attenuated (e.g., the Atto-Arc system from Carl Zeiss or metal halide burners, e.g., from Nikon), or increase magnification slightly.
Reducing light intensity by closing diaphragms is not recommended as it reduces resolution. In AVEC microscopy, if the one-ninth setting yields too much light for the camera, this could be reduced by setting the compensator or Wollaston prism to a position of less than 20°. Although widely used, this does somewhat compromise image quality, especially if settings of less than 15° or 10° are used (Allen et al., 1981a,b; Hansen et al., 1988).
Live cells from tissue cultures should preferentially be grown on a cover glass. The specimen's region of interest should be close to the cover glass surface, where the best image is obtained. If the highest magnifications are intended, it may be that the optics can be adjusted for Köhler illumination only at this surface and a few tens of micrometers below (upright microscope), as high-magnification objectives are usually designed for best optical imaging of objects only near a distance of 170µm from the front element, i.e., 10- 20µm from the inner surface of a regular cover glass (No. I glasses, approximately 170µm thick). To identify objectives for which this distance and the presence of a 170-µm-thick cover glass is critical, look for the "0.17" engraved or refer to the manufacturer's data sheet. If thick cells or other extended specimens must be observed with such objectives, No. 0 cover glasses (80-120µm thick) and/or thinner glass slides (0.9 instead of 1 mm) (both available from O. Kindler GmbH or Scholt North America) may have to be used instead of the regular ones to allow the setting up of Köhler illumination with oil immersion of both condenser and objective lens and to permit a greater working distance within the specimen. The use of a slide preparation made of two cover glasses mounted in a frame as depicted in Fig. 1 would serve the same purpose.
Note, however, that if the oil immersion objectives are focusing through water rather than through glass and oil only, spherical aberration will be introduced. The use of monochromatic illumination or of immersion oils of different refractive indices (R. P. Cargille Laboratories) is recommended to overcome this problem. Alternatively, for imaging deep within an aqueous sample, water immersion objectives must be used. Special water immersion objectives have become available from major manufacturers for confocal microscopy. They combine high numerical aperture (e.g., 60x, NA 1.2) with correction for extreme working distances of up to 220 µm below the cover glass (Brenner, 1994).
If working with an inverted microscope, the specimen slide has to be inverted for fitting to the microscope with the cover glass underneath. With most microscope stages, this will interfere with the VALAP sealant, and flat positioning of the slide will not be possible. Instead, use of a metal frame the size of a regular slide and 0.8 to 1mm thick to hold a sandwich of two cover glasses of dissimilar sizes separated by tape or cover glass spacers is recommended (Schnapp, 1986; Weiss et al., 1999, 1990) (Fig. 1).
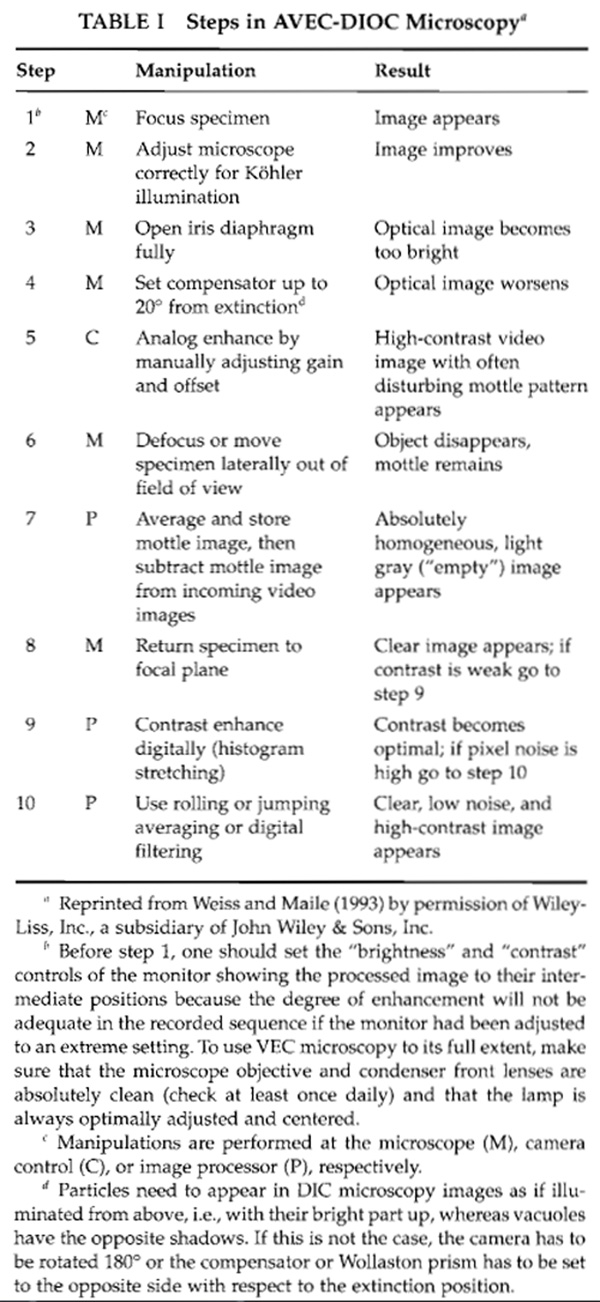
![FIGURE 1 Slide preparation suitable for highest magnification VEC microscopy with upright and inverted microscopes and for superfusion of live cells or suspension specimens: (1) adhesive tape attaching the cover glass to the metal frame (7); (2) drop of medium to replace the original medium; (3) VALAP sealant; (4) small cover glasses or thin adhesive tape as spacers (e.g., double-sticky Scotch Tape); (5) carrier cover glass [e.g., No. 2 (extra thick) 24 × 50 or 60 mm]; (6) filter paper wick to induce medium flow (arrow); and (7) metal frame of the size of regular microscope slides made of brass or aluminum 0.8 to 1mm thick and absolutely flat. For use with inverted microscopes the preparation should be mounted on the lower side of the carrier glass (5). Reproduced from Weiss et al. (1990) with permission by Plenum Press.](images/v3_pa_s02_c06_f01.jpg) |
| FIGURE 1 Slide preparation suitable for highest magnification VEC microscopy with upright and inverted microscopes and for superfusion of live cells or suspension specimens: (1) adhesive tape attaching the cover glass to the metal frame (7); (2) drop of medium to replace the original medium; (3) VALAP sealant; (4) small cover glasses or thin adhesive tape as spacers (e.g., double-sticky Scotch Tape); (5) carrier cover glass [e.g., No. 2 (extra thick) 24 × 50 or 60 mm]; (6) filter paper wick to induce medium flow (arrow); and (7) metal frame of the size of regular microscope slides made of brass or aluminum 0.8 to 1mm thick and absolutely flat. For use with inverted microscopes the preparation should be mounted on the lower side of the carrier glass (5). Reproduced from Weiss et al. (1990) with permission by Plenum Press. |
Step 1: Focusing the Specimen
We find the specimen preferably by looking through the oculars or, alternatively, by looking at reduced magnification at the monitor. Only if the entire specimen consists of subresolution size material (density gradient fractions, microtubule suspensions, unstained EM sections) will it be difficult to find the specimen plane. Use a relatively dark setting of the condenser diaphragm and/or polarizers or prisms and look for contaminating larger particles. If there are none, routinely apply a fingerprint to one corner of the specimen side of the cover glass and use this for focusing.
Step 2: Adjusting Köhler Illumination
After finding a coarse setting for the illumination, the desired plane for the specimen is selected exactly. Then the condenser is finely adjusted, but now in relation to the image on the monitor (make sure the light is reduced to avoid damage of the camera!). The field diaphragm must be centered on the monitor and opened until it becomes just invisible. If the field diaphragm is opened too much, most microscopecamera adapter tubes or high-power projectives and oculars will create a very annoying central hot spot. If this persists at the adjustment for proper illumination, closing the projective diaphragm or inserting a selfmade diaphragm cutting the peripheral light at the microscope exit usually helps. Note that at the high magnifications and numerical apertures used here, Köhler illumination has to be readjusted once the focus is changed more than a few micrometers.
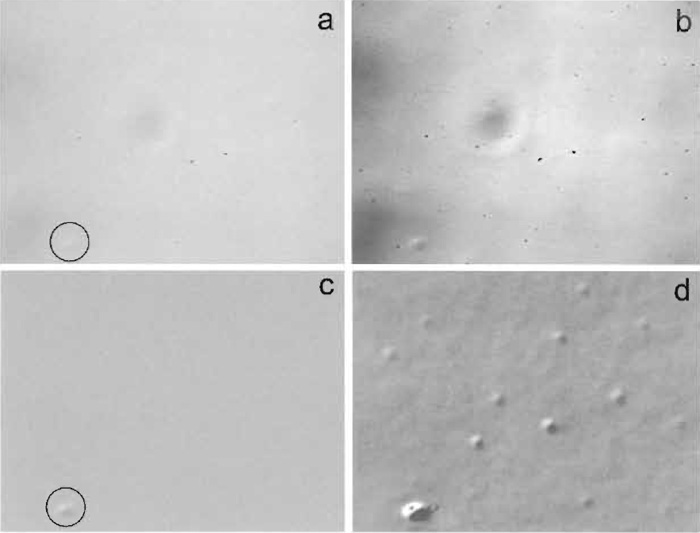 |
| FIGURE 2 VEC microscopy of a specimen with very weak contrast. The steps of image generation and contrast enhancement in VEC microscopy are demonstrated using subresolution size latex beads (50 nm diameter). (a and b) Analog contrast enhancement. (c and d) Digital background subtraction and digital contrast enhancement. (a) In focus, not enhanced; only the large aggregate of a size above the limit of resolution is visible (circle); (b) in focus, analog-enhanced; the mottle becomes annoying; (c) in focus, mottle subtracted; (d) the same digitally enhanced. Microscope: Reichert-Leica Polyvar; image processor: Hamamatsu Argus 10; width of frames: 22 µm. |
Open the condenser diaphragm fully in order to utilize the highest possible numerical aperture to obtain highest resolution. Also, any iris diaphragm of the objective should be opened fully. The result of opening the condenser diaphragm will usually be that the optical image worsens because it becomes too bright and flat for the eye. This setting will result in a small depth of focus, especially with DIC (optical sections of 0.3 µm or less with 100x NA 1.4 oil objectives). If collection of information from a large depth of focus is desired (e.g., viewing very dilute suspensions) and resolution can be sacrificed, the condenser diaphragm may be closed down as desired.
Be careful to protect the camera from too high light intensity prior to this step. However, at this point you have to make sure that the camera receives the proper amount of light to work near its saturation end. Some manufacturers have red and green control lamps built in to indicate this. In any case you should see a moderately modulated image on the monitor, whereas a very flat or no image indicates insufficient light. In this case, follow the recommendations on how light intensity can be increased (see Section III). If the illumination has to be reduced to protect the camera, this should not be done by closing diaphragms, as this compromises resolution (see Section III).
Step 4: Setting the Compensator (Polarized Light Techniques Only)
Set the compensator or main prism (AVEC-DIC) or the polarizer (AVEC-POL) to about 1/9 λ, i.e., oneninth of a wavelength. The optical image, i.e., that seen in the oculars, will disappear due to excessive stray light.
If you do not have such a calibrated system, first determine the distance between extinction (0° and maximum brightness (90° or one-half of a wavelength) by moving the adjustable Wollaston prism and then estimate and select approximately the ninth of a wave or 20° position. Many microscopes equipped with DIC for biological applications do not allow a phase shift of 90° and some may not even allow 20° because for observation by eye, phase shifts of a few degrees already yield good contrast. Microscope manufacturers will, however, have the proper parts in their mineralogy programs.
At this point you have to again make sure that the camera is protected from excessive light but receives enough light to work near its saturation end. Ideally this should be near the one-ninth setting. Light adjustment should be done as explained in Section III. Opening the compensator or the crossed polarizers beyond 20° although perhaps leading to a better saturation of the camera, will hardly improve the resolution of DIC or polarization images and will yield a poor image resembling the bright-field type.
Step 5: Analog Enhancement
Increase the gain on the camera to obtain good contrast. Then apply offset (pedestal). Always stop before you lose parts of the image that become too dark or too bright. Repeat this procedure several times, if necessary and helpful. Make sure that the monitor for watching the changes is not set to extreme contrast or brightness and is terminated properly (75Ω setting). Analog enhancement improves the contrast of the specimen, but unfortunately also emphasizes dust particles, uneven illumination, and optical imperfections. These artifacts, called "mottle," are superimposed on the image of the specimen and may, in some cases, partially or totally obscure it (Fig. 2). Disturbing contributions from fixed pattern noise (mottle) or excessive amounts in unevenness of illumination (Fig. 2b) can, however, be tolerated if digital processing is performed later (Fig. 2c and d).
If digital processing is not possible, stop enhancement just before mottle or uneven illumination becomes annoying. Optimal adjustment of the lamp and thorough cleaning of the inner optical surfaces of the microscope, oculars, and camera lens usually result in images that allow the application of considerably higher analog contrast enhancement. Apply analog shading correction and other types of analog image improvement if your camera control unit offers these features.
Step 6: Finding a "Background" Scene
Try removing the specimen laterally out of the field of view or (when using DIC) defocus to render it just invisible (preferably toward the cover glass). The result is an image containing only the imperfections of your microscope system (mottle pattern). This step may not be satisfactory, however, with such techniques as phase contrast or bright field.
Step 7: Background (Mottle) Subtraction
Freeze, i.e., store, the mottle image, preferably averaged over several frames, and subtract it from all incoming video frames.
When returning the specimen to the focal plane, you should see an absolutely even and clean image, which may, however, be weak in contrast (Fig. 2c). If there are regions "missing" that are gray and flat, there is too much contrast in the mottle of the raw image to be subtracted properly. Reduce gain, adjust offset, and repeat the procedure.
Step 9: Digital Enhancement
This is done in a similar manner to step 5. Alternate between stretching a selected range of gray levels (setting "width") and shifting the image obtained up and down the scale of gray levels (setting "level" or "lower level") until a pleasing result is found (Fig. 2d). If available on your equipment, display the gray level histogram and select the upper and lower limits, which are to be defined as bright white and saturated black, respectively. If the image is noisy (pixel noise), go to step 10.
Step 10: Temporal Filtering (Frame Averaging)
Use an averaging function in a rolling (recursive filtering) or jumping mode over two or four frames. This will still allow the observation of fairly rapid movements in your specimen, whereas very fast motions and noise due to pixel fluctuations will be averaged out. Averaging over a large number of frames will filter out all undesired motion and may be used with a fixed specimen or in order to remove, for example, distracting Brownian motion of small particles in suspension. The image will then contain the immobile parts of the object exclusively.
Step 11: Spatial Filtering
A large number of digital procedures for spatial filtering are available that can be used to reduce noise, to enhance edges of objects, or to reduce shading (Shotton, 1993; Russ, 1995). Some image processors offer such filters at video rate so that live sequences can be accentuated by filtering prior to recording.
Step 12: Printing Pictures
For publication purposes, pictures may be photographed or filmed off the video screen by observing special procedures and hints to remove video lines as discussed in detail by Inoué and Spring (1997) or Weiss et al. (1999). Alternatively, video printers are available with near-photographic or, at higher cost, with true pho-tographic quality that can be directly connected to the analog video output. Very good results are also obtained if all analog and photographic steps for printing can be avoided and the selected frame is directly transferred in digital form to a computer and from there to a high-resolution printer.
Unlike in EM images, which truly resolve the submicroscopic objects depicted, the sizes of objects seen by AVEC-DIC microscopy may not necessarily reflect their real size. When AVEC-DIC is used, the limit of true resolution is shifted to almost one-half of that obtained conventionally. Objects smaller than the limit of resolution, i.e., 100-250nm, depending on the optics and the wavelength of light used, are inflated by diffraction to the size of the resolution limit. The orientation of birefringent objects may also somewhat affect their apparent thickness if they are oriented at angles very close to 45° or 135°. Whereas the size of the image does not enable a decision on whether one or several objects of a size smaller than the limit of resolution are pre-sent, the contrast sometimes permits this judgment to be made. A pair of microtubules would, for example, have the same thickness as a single one, but the contrast would be about twice as high. If large numbers of subresolution objects are separated by distances of less than 200 nm from each other (e.g., microtubules in concentrated suspensions or vesicles in a synapse), they will remain invisible, but they will be depicted clearly if they are separated by more than the resolution limit.
Also remember that if in-focus subtraction or averaging over time is used, the immobile or the moving parts of the specimen, respectively, may have been completely removed from the image.
Allen, R. D., Allen, N. S., and Travis, J. L. (1981a). Video-enhanced contrast, differential interference contrast (AVEC-DIC) microscopy: A new method capable of analyzing microeubule-relaeed motility in the reeiculopodial network of Atlogromia Iaticollaris. Cell Molil. 1, 291-302.
Allen, R. D., Travis, J. L., Allen, N. S., and Yilmaz, H. (1981b). Videoenhanced contrast polarization (AVEC-POL) microscopy: A new method applied to the detection of birefringence in the motile reticulopodial network of Allogromia lalicollaris. Cell Motil. 1, 275-288.
Bennett, H. S. (1950). Methods applicable to the study of both fresh and fixed materials: The microscopical investigation of biological materials with polarized light. In "Handbook of Microscopical Technique" (C. E. McClung, ed.), pp. 591-677. Harper and Row (Hoeber), New York.
Brenner, M. (1994). Imaging dynamic events in living tissue using water immersion objectives. Am. Lab. April, 3844 (also as Technical Bulletin from Nikon Inc. Melville, NJ).
Dodt, H. U., and Zieglgäinsberger, W. (1990). Visualizing unstained neurons in living brain slices by infrared DIC-video microscopy. Brain Res. 537, 333-336.
Foskett, J. K. (1993). Simultaneous differential interference contrast and quantitative low-light fluorescence video imaging of cell function. In "Optical Microscopy: Emerging Methods and Applications" (B. Herman and J. J. Lemasters, eds.), pp. 237-261. Academic Press, New York.
Hansen, E. W., Conchello, J. A., and Allen, R. D. (1988). Restoring image quality in the polarizing microscope: Analysis of the Allen video-enhanced contrast method. J. Opt. Soc. Am. AS, 1836- 1847.
Inoué, S. (1961). Polarization microscope. In "The Encyclopedia of Microscopy" (G. L. Clark, ed.), pp. 480-485. Reinhold, New York.
Inoué, S. (1981). Video image processing greatly enhances contrast, quality, and speed in polarization-based microscopy. J. Cell Biol. 89, 346-356.
Inoué, S., and Spring, K. R. (1997). "Video Microscopy", 2 nd Ed., Plenum Press, New York, 741 pp.
Inoué, S. (1989). Imaging of unresolved objects, superresolution, and precision of distance measurement with video microscopy. In "Methods in Cell Biology" (D. L. Taylor and Y.-L. Wang, eds.), Vol. 30, pp. 85-112. Academic Press, New York.
Kachar, B. (1985). Asymmetric illumination contrast: A method of image formation for video light microscopy. Science 277, 766-768.
Russ, J. C. (1995). "The Image Processing Handbook," 2nd Ed. CRC Press, Boca Raton, FL.
Schnapp, B. J. (1986). Viewing single microtubules by video light microscopy. Methods Enzymol. 134, 561-573.
Shotton, D. (1993). An introduction to digital image processing and image display in electronic light microscopy. In "Electronic light Microscopy" (D. Shotton, ed.), pp. 39-70. Wiley-Liss, New York.
Weiss, D. G., and Maile, W. (1993). Principles, practice, and applications of video-enhanced contrast microscopy. In "Electronic Light Microscopy" (D. Shotton, ed.), pp. 105-140. Wiley-Liss, New York.
Weiss, D. G., Maile, W., Wick, R. A., and Steffen, W. (1999). Video microscopy. In "Light Microscopy in Biology. A Practical Approach" 2nd Ed. (A. J. Lacey, ed.), pp. 73-149. Oxford University Press, Oxford.
Weiss, D. G., Meyer, M., and Langford, G. M. (1990). Studying axoplasmic transport by video microscopy and using the squid giant axon as a model system. In "Squid as Experimental Animals" (D. L. Gilbert, W. J. Adelman, Jr., and J. M. Arnold, eds.), pp. 303-321. Plenum Press, New York.
Support our developers




