Cleaving C—C Bonds With the Help of Coenzymes
The breakdown of fats, sugars, and other foods as well as the synthesis of body constituents depends upon numerous processes of making and breaking chemical bonds. The cutting and forming of C—C bonds is especially challenging. Enzymes can utilize chemical groupings of an acidic or basic character that are present in the amino acids from which the proteins are made. The acidic −COOH, imidazolium (from histidine), and −NH3+ groups serve as proton donors, and the unprotonated forms of these same groups, as proton acceptors. These groups facilitate cleavage and formation of O—H, N—H, and C—H bonds. Certain C—C bonds, e.g., those that are one atom removed from a carbonyl group, can also be broken by proteins using only their own catalytic acid–base groups. This is illustrated in Fig. 13. The carbonyl group provides an electronaccepting center into which electrons can flow temporarily as the C—C bond is broken. Both the cleavage of a β oxo alcohol and a β oxoacid (decarboxylation) are illustrated. In some instances the carboxyl (—COOH) group of a substrate can serve as an electron acceptor. However, the reactivity toward bond cleavage is much higher in a thioester such as that formed from acetyl-coenzyme A (Fig. 10). This high reactivity accounts for one function of coenzyme A. For example, coenzyme A permits the cleavage, by a reverse Claisen condensation, of the fatty acid chain during the β oxidation of fatty acids (Fig. 12, step e). However, not all C—C bonds can be broken using only the chemical groupings of the proteins or of coenzyme A.Participation of thiamin diphosphate or pyridoxal phosphate is required for many other C—C bond cleavages. Thiamin diphosphate enables cleavage of an α oxoacid as indicated in Fig. 13. A characteristic of thiamin diphosphate is that, when bound correctly into an active site, it can lose a proton from its 5-membered thiazolium ring to form the dipolar ionic “ylid” structure shown in Fig. 13. This can add to the carbonyl group of an α oxoacid or an α oxoalcohol to form a covalent compound (adduct) in which the double bond of the thiazolium ring provides the necessary electron-accepting center. The positive change on the nitrogen atom of the ring assists in initiating the chain cleavage. These thiamin-dependent cleavage reactions are found in virtually every major metabolic pathway in higher organisms and in most bacteria. For example, the acetyl-CoA that is generated by β oxidation of fatty acids (Fig. 12) enters the citric acid cycle where the two carbon atoms of the acetyl group are converted to CO2. One essential step in the cycle requires thiamin diphosphate. It is hard to imagine how such metabolic cycles could be organized without thiamin diphosphate.
Pyridoxal phosphate, sometimes in collaboration with pyridoxamine phosphate, participates in dozens of different reactions of amino acids, the building blocks of proteins. These reactions involve both the biological synthesis of amino acids and the breakdown of amino acids, e.g., of excess amino acids in the human diet. For these reactions, the PLP is held in place by the enzyme in a location adjoining the binding site for the specific amino acid substrate. In this site an amino group of a protein side chain (a lysine side chain; Protein −NH2) forms a Schiff base linkage in which the carbonyl (C=O) group of PLP is converted to a Schiff base linkage (C=N—Protein) similar to that present in the PLP Schiff base drawn in Fig. 14. This is the “resting form” of the enzyme. Then, in a two-step process, the amino group of the substrate adds to the C=N bond and displaces (eliminates) the Protein −NH2 group to form the substrate Schiff base that is shown in generalized form in Fig. 14. In this Schiff base, one of the three bonds (a, b, c,) may be broken. This is illustrated for cleavage a, removal of a hydrogen atom as H+ by a catalytic group of the protein. The small arrows beside the structure indicate the manner in which the pyridine ring of the coenzyme, with a proton bound to the nitrogen atom of its ring, serves as an electron acceptor in much the same way as does thiamin diphosphate (Fig. 13). The structure resulting from removal of the α-proton of the PLP Schiff base is variously known as a “quinonoid” or “carbanionic” intermediate. Depending upon the specificity of the enzyme in whose site it is formed, this intermediate may react in several ways. In a bacterial racemase a proton may be returned to the α-carbon atom from which it was removed but without stereospecificity, i.e., into either of two positions relative to the other groups surrounding the α-carbon. Some racemases are used by bacteria to convert the stereoisomer known as L-alanine into the less common “unnatural” D-alanine. The latter is incorporated into the bacterial cell wall and helps provide protection to the bacteria against attack by hydrolytic enzymes.
A second mode of reaction of the quinonoid-carbanionic intermediate is utilizedbyplants whichsynthesize an enzyme that acts on the amino acid S-adenosylmethionine to form a cyclic three-membered ring compound aminocyclopropane carboxylic acid. This is a major plant hormone. In a third type of reaction a proton is added back to the coenzyme itself (see Fig. 14) to form what is called a ketimine (not illustrated). This is a Schiff base of pyridoxamine phosphate (PMP, Fig. 5) with an α-oxoacid and is an essential intermediate compound in the important process of transamination (Fig. 14). This process is utilized by all living organisms both in the synthesis of amino acids and in the breakdown of excesses of amino acids. The human body forms several amino acids via transamination. As shown in Fig. 15, this is a reversible sequence involving a cyclic interconversion of PLP and PMP in reaction steps of the type illustrated in Fig. 14.
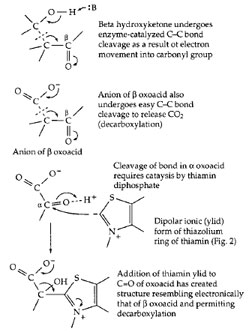 |
| Figure 13 Activation of C—C bond cleavage by adjacent carbonyl group (top) and by formation of adduct with thiamin diphosphate (bottom). |
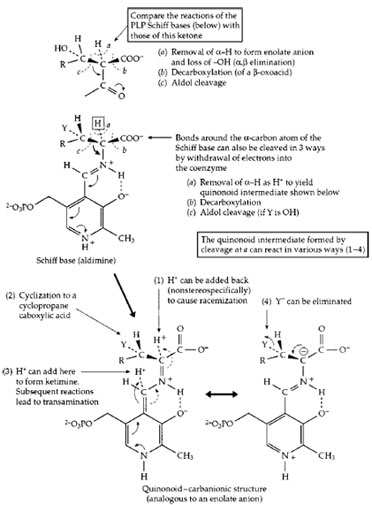 |
| Figure 14 The action of pyridoxal phosphate in initiating catalysis of numerous reactions of α-amino acids. Completion of the various reactions requires a large variety of different enzyme proteins. |
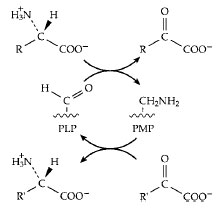 |
| Figure 15 The transamination reaction by which amino groups are transferred from one carbon skeleton (in the form of an α oxoacid) to another to form or to degrade an amino acid. |
A third coenzyme that is involved in C—C bond cleavage and formation is the vitamin B12 derivative 5´-deoxyadenosylcobalamin (Fig. 7). In this compound the cobalt–carbon bond is easily cleaved to form a free radical which, in turn, facilitates C—C bond cleavage in the substrate. The details, which are still under study, have beenomitted, but Fig. 16 shows a general reaction in which group X is often attached via a C—C bond which is broken. The net result is that a hydrogen atom trades places with group X. These rearrangement reactions, which cannot be catalyzed by proteins alone or by other coenzymes, are quite numerous in various bacteria. However, only one of them occurs in human cells. That is the conversion of methylmalonyl-CoA to succinyl-CoA, the reverse of the succinyl-CoA mutase reaction as drawn in the lower section of Fig. 16. The reaction is essential to the metabolism of propionyl-CoA as is indicated at the bottom of Fig. 12. Propionyl-CoA is carboxylated at the site marked by an arrow in Fig. 17 to form methylmalonyl- CoA. This compound must be isomerized by the vitamin B12-dependent mutase to form succinyl-CoA which can be oxidized to CO2 in the body’s central metabolic pathways. Lack of the mutase is fatal.
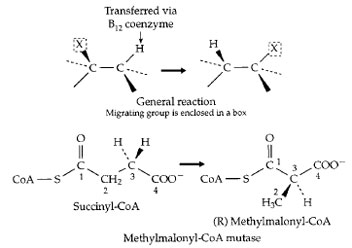 |
| Figure 16 (Top) A family of rearrangement reactions that depend upon free radical formation involving an enzyme-bound form of the vitamin B12 coenzyme 5´ -deoxyadenosylcobalamin (Fig. 7). The rearrangement of (R) methylmalonyl-CoA to succinyl-CoA (the opposite of the reaction shown here) is one of the two essential vitamin B12-dependent reactions in the human body, and plays an important role in fatty acid oxidation, as is indicated in Fig. 12. |
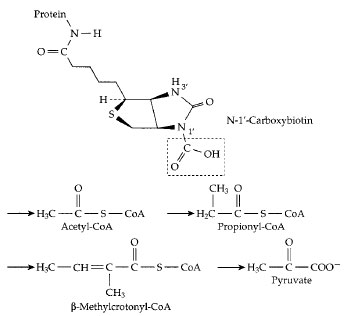 |
| Figure 17 The carboxyl carrier function of biotin. A molecule of activated CO2 is carried as —COOH bonded to N−1´ of biotin, which is covalently attached (as in Fig. 11) to an appropriate protein. Below this structure the sites of four different metabolic intermediates that receive activated CO2 from carboxybiotin are marked by arrows. In each case, either the thioester linkage to coenzyme A or another adjacent carbonyl group activates a hydrogen atom which dissociates as H+, leaving a negatively charged site which accepts the CO2 by direct transfer from carboxybiotin. Carboxylation of propionyl-CoA in the human body is an essential step in degradation of branched chain and odd chainlength fatty acids (Fig. 12). The resulting methylmalonyl-CoA is converted to succinyl-CoA, the reverse of the reaction shown in Fig. 16. |
Support our developers




