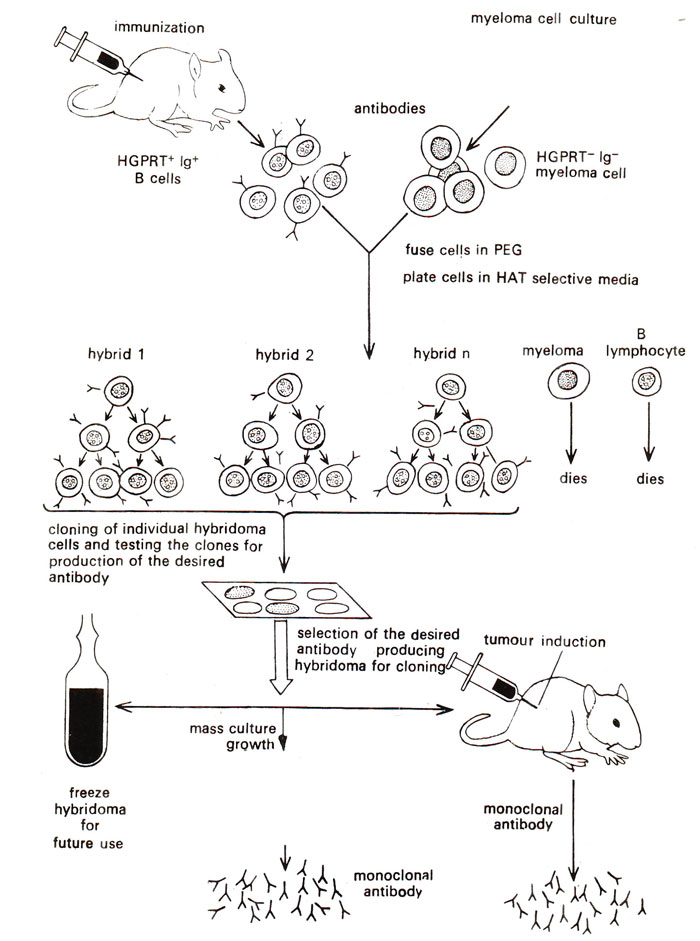
Fig. 43.1. Procedure for making and selecting hybridomas for the production of monoclonal antibodies.
Monoclonal antibodies can be produced in specialized cells through a technique now popularly known as
hybridoma technology. This technology was discovered in 1975 by two scientists,
Georges Kohler of West Germany and
Cesal Milstein of Argentina (now working in U.K.), who jointly with
Niels Jerne of Denmark (now working in Germany) were awarded the
1984 Nobel Prize for Physiology and Medicine. The term
hybridoma is applied to fused cells resulting due to fusion of following two types of cells : (i) antibody producing
lymphocyte cell (e.g. a spleen cell of mouse immunized with red blood cells from sheep), and (ii) a single
myeloma cell (bone marrow tumour cell) which is capable of multiplying indefinitely. These fused hybrid cells or hybridoma have the antibody producing capability inherited from lymphocytes and have the ability to grow continuously (immortal) like malignant cancer cells. Following steps are involved in the production of monoclonal antibodies using hybridoma technology (Fig. 43.1):
(i) Immunize a rabbit through repeated injection of a specific antigen for the production of specific antibody, facilitated due to proliferation of the desired B cells, (ii) Produce tumours in a mouse or a rabbit, (iii) From the above two types of animals, culture separately spleen cells (spleen cells are rich in B cells and T cells) that produce specific antibodies and myeloma cells that produce tumours (the myeloma cell line used, is unusual in two ways; it has stopped synthesizing antibodies and it is a mutant called HGPRT
-, that can not synthesize the enzyme
hypoxanthine-guanine phospho-ribosyl transferase or
HGPRT). (iv) Induce fusion of spleen cells to myeloma cells, using
polyethylene glycol (PEG), to produce hybridoma; the hybrid cells are grown in selective
hypoxanthine aminopterin thymidine (HAT) medium. HAT medium contains a drug aminopterin, which blocks one pathway for nucleotide synthesis, making the cells dependent on another pathway that needs HGPRT enzyme absent in myeloma cells. Therefore myeloma cells that do not fuse with B cells will die since they are HGPRT
-. B cells that do not fuse will also die because they lack tumorigenic property of immortal growth. Therefore HAT medium allows selection of hybridoma cells, which inherit HGPRT gene from B cells and tumorigenic property from myeloma cells, (v) Select the desired hybridoma for cloning and antibody production; this is facilitated by preparing single cell colonies that will grow and can be used for screening of antibody producing hybridomas; only one in several hundred cell hybrids will produce antibodies of the desired specificity; (vi) Culture selected hybridoma cells for the production of monoclonal antibodies in large quantity; these hybridoma cells may be frozen for future use and may also be injected in the body of the animal so that antibodies will be produced in the body and can be recovered later from the body fluid (Fig. 43.1).
Fig. 43.1. Procedure for making and selecting hybridomas for the production of monoclonal antibodies.
Improvements in hybridoma technology
Considerable efforts during the last 10-15 years (1980s and 1990s) have been made to improve the yield of monoclonal antibodies using hybridoma technology. These efforts included the following : (i) As indicated above, cell fusions were facilitated through the use of
polyethylene glycol (PEG). (ii) A continuous cell line
(SP2/0) was used as a fusion partner for the antibody-producing B cells, (iii)
Feeder layers consisting of extra cells to feed newly formed hybridomas were used for optimal growth and hybridoma production; the most common feeder layers consisted of (i) murine peritoneal cells, (ii) macrophages derived from mouse, rat or guinea pigs, (iii) extra non-immunized spleen cells, (iv) human fibroblasts, human peripheral blood monocytes or thymus cells. These feeder cells had some limitations like depletion of nutrients meant for hybridoma and contamination, so that other sources of hybridoma growth factors
(HGF) like
interleukin-6 (IL-6) derived from human cells were used.
Purification of antibodies
Monoclonal antibodies may need to be purified before they are used for a variety of purposes. Before final purification, the cultures (whether cell cultures or bacterial cultures with cloned genes, as described later) may be subjected to cell fractionation for enrichment of the antibody protein. In
E. coli, the antibodies may be secreted in the periplasm, which may be used for enrichment of antibody, so that further purification is simplified. Alternatively the antibodies may be purified from cell homogenate or cell debris obtained from the medium. Antibodies can be purified by any one of the following two techniques (i) ion-exchange chromatography; (ii) antigen-affinity chromatography.
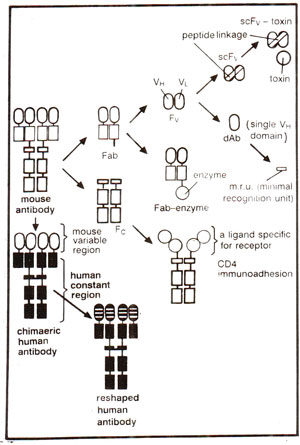
Fig. 43.2. Genetically engineered antibodies and fragments (see Fig. 37.13 for different fragments).
Antibody Engineering and Genetic Manipulations
Although hybridoma technology enables us to obtain monoclonal antibodies or homogenous antibodies of predefined specificity, it can not allow production of novel antibodies of desired specificity. Such novel antibodies can be obtained by redesigning antibodies through the application of gene technology, an area of research which has revolutionized the potential of monoclonal antibodies (for structure of antibody). For application of gene technology, an initial but crucial problem was to find a convenient way for expression of the cloned antibody genes into proteins (antibodies). For this purpose expression could be obtained in (i) mammalian cells and (ii) bacterial cells, particularly
E. coli. Initially, antibody genes were taken from hybridomas, cloned into plasmid vectors, and expressed as complete antibodies in mammalian cells. However, later genes for different small fragments of antibodies (e.g.
Fv, Fab, Fc or even single chain
Fv =
scFv) could be cloned and expressed in bacteria (Fig. 43.2). Expression in mammalian cells becomes important, when glycosylation of antibodies is desired. However, lack of glycosylation does not influence antigen binding capacity of Fv or Fab fragments or of complete antibody, although it does affect some of the effector functions attributed to Fc fragment.
Therefore for industrial production of antibodies, glycosylation is immaterial and
E. coli can be used as a useful expression system. Following are some of the advantages of the use of
E. coli : (i) fast growth; (ii) simple fermentation to allow large scale production in fermenters; (iii) fragments can be produced by
Fv or
Fab genes and not from complete antibody through proteolysis; these small fragments can be used for a variety of'studies on structure (through NMR and crytallography) and function; (iv) antibodies in the form of bifunctional molecules can be produced (e.g. antigen binding + toxin); (v) screening for binding activity or catalytic activity can be done on bacterial colonies or phage plaques, without isolating antibodies; this screening procedure can be effectively used for screening antibody gene libraries (see later).
Fig. 43.2. Genetically engineered antibodies and fragments (see Fig. 37.13 for different fragments).




