Fractionation of cellular components
The living cell contains a number of subcellular particles. Much of our knowledge of the functions and enzymic properties of these particles has been derived from a study of pure preparation of them. The fractionation of plant cells involves two distinct phases: disruption of the tissue or cells in a suitable medium and the subsequent separation of the subcellular particles, by differential centrifugation which exploits differences in their size and density. This procedure results in rather crude subcellular fractions which are enriched with one particular component and are by no means pure. These fractions are then purified by density gradient centrifugation. The identity of the type of organelle isolated in a particular fraction is checked by microscopic examination and by looking for the presence of a compound or enzyme characteristics of it; the latter is known as marker.Disruption of Cells
Since the plant cell wall is tough, disruption is done mostly by mechanical means. The most common method employs a blender whose high-speed blades exert large shearing forces on the cells. The tissue is immersed in an equal weight of homogenizing medium and blended for 0.5-2 min at full speed. Unfortunately this procedure is far severe for the delicate cellular components and may be partly damaged. Probably the gentlest procedure of all is hand-grinding of chopped pieces of sample with a pestle and mortar in an equal volume of homogenizing medium sometimes with a little acid-washed sand to act as an abrasive. All these cell disruption procedures are carried out rapidly at 2-4°C to minimize autolytic changes.
Homogenization Media
The ideal homogenization medium should be capable of maintaining the morphological and functional integrity of the organelles. Homogenization media usually contain:
(i) 0.3M mannitol so as to be isotonic with the cytosol;
(ii) a pH 7-8 buffer (often Tris) at about 50mM concentration to neutralize the acidic vacuolar contents;
(iii) a sulphydryl compound (dithiothreitol or mercaptoethanol) at about 10mM concentration to minimize the inactivation of enzymes;
(iv) Mg2+ at about 10mM concentration to keep ribosomes intact;
(v) Ca2+ at about 1mM concentration to prevent the clumping of nuclei; and
(vi) polyvinylpyrrolidone and bovine serum albumin (0.1-0.2%) to precipitate out the tannins and phenolics.
Purification
The cell homogenate is fractionated using differential centrifugation into at least five major fractions namely nuclei, chloroplasts, mitochondria, microsomes and supernatant. These fractions are however impure and can be purified by a number of techniques such as centrifugation, phase separation, electrophoresis and specific adsorption of these components. Centrifugation remains the most generally used procedure. Again, a number of centrifugation media such as sucrose, ficoll and metrizamide are used either on a linear or nonlinear density gradients.
A generalized procedure for separating a tissue homogenate into crude fractions enriched in a particular cell component is given in the flow-sheet.
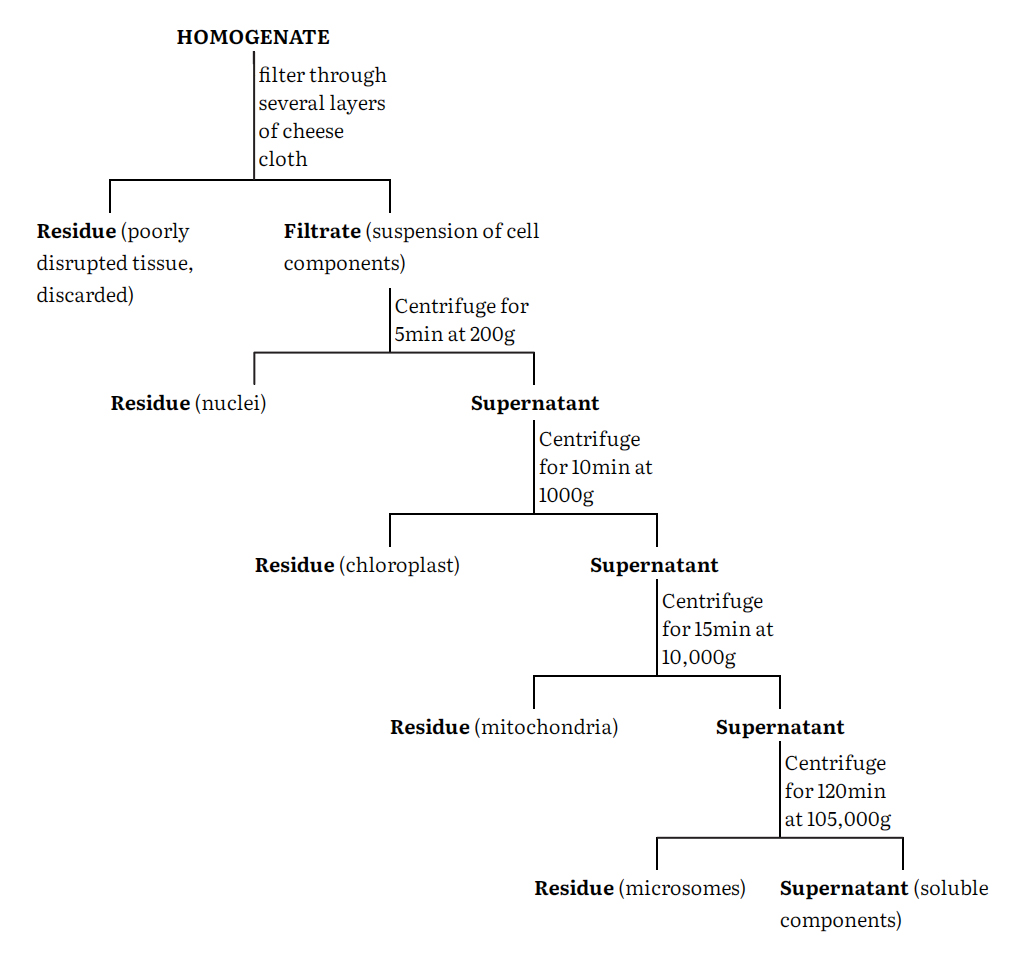
The particular fraction is resuspended in homogenization medium and then carefully layered onto the top of a sucrose gradient in a centrifuge tube. The gradient is made by successively adding layers of sucrose solution of decreasing concentration (and therefore density) to the centrifuge tube so gently that they do not mix to any great extent. The tube may be allowed to stand at 2-4°C for about 30 min to allow diffusion to smooth the steps in the gradient. After the resuspended cell fraction has been added, the tube is centrifuged in a swinging bucket rotor. Depending upon the nature of gradient and the length of the centrifugation period different particles separate and band at zones where their density equal that of sucrose solution. After centrifugation, these bands are carefully pipetted out separately as pure fractions and further analyzed.
Support our developers




