Immunoelectron Microscopy with Lowicryl Resins
I. INTRODUCTIONLowicryl resins (Carlemalm et al., 1982) are well suited for electron microscope immunocytochemistry of formaldehyde or formaldehyde/glutaraldehydefixed tissues. They provide both good ultrastructural preservation and preserve to a large extent tissue antigenicity. Embedding in acrylic resins is carried out at low temperature either by progressively lowering the temperature during dehydration and resin infiltration or by cryofixation followed by freeze substitution and UV polymerization at low temperature (Carlemalm et al., 1985; Schwarz and Humbel, 1989; Sitte et al., 1989). Thin Lowicryl sections can then be cut on an ultramicrotome at room temperature and labeled with the appropriate primary antibody, which can then be detected with a secondary antibody (or protein A) coupled to colloidal gold particles. A number of labeling principles and protocols have been developed and discussed (Griffiths, 1993; Larsson, 1988; Newman and Hobot, 1993; Roth, 1986; Schwarz and Humbel, 1989). The protocol given here represents a standard procedure we have applied in our laboratory for the localization of several different antigens at the ultrastructural level (Maunsbach and Afzelius, 1998).
Unstained sections of biological tissues show low contrast when examined in a transmission electron microscope. Almost invariably immunogold-labeled sections are therefore contrasted, or stained, by interaction with one or more solutions containing salts of heavy metals, such as uranyl acetate (Watson, 1958) and/or lead citrate (Reynolds, 1963). The staining is unspecific from a chemical point of view, but serves to enhance the contrast of cellular components in the microscope. In immunogold electron microscopy, section staining is important in order to determine the precise relationship between colloidal gold particles and tissue fine structure.
II. FREEZE SUBSTITUTION IN LOWICRYL HM20
A. Materials and Instrumentation
Liquid nitrogen
Lowicryl HM20 kit (Agar Scientific Ltd) containing HM20 resin (monomer E), HM20 cross-linker (D), HM20 initiator (C)
Water-free methanol (CH3OH, Merck 106007)
Silica gel beads (Sigma S7651)
Sodium chloride (NaCl, Merck 106404)
Sodium dihydrogen phosphate monohydrate (NaH2PO4·H2O, Merck 106346) Disodium hydrogen phosphate dihydrate (Na2HPO4·2H2O, Merck 106580)
Sucrose (C12H22O11, BHD, Analar 102745c)
Uranyl acetate dihydrate [(CH3COO)2UO2·2H2O, Polysciences 21477]
Fine forceps with cold-insulated shaft
Polyethylene capsules with pyramid shape and hinged lids for low-temperature embedding (BEEM capsules G360-1, Agar Scientific Ltd., Figs.1a and 1b)
Disposable beaker for mixing resin
Polyethylene Pasteur pipettes with extended fine tips (Figs. 1a and 1b)
Freeze-substitution apparatus with temperature regulation between -85 and 0°C
Freeze-substitution unit (Leica EM AFS, Leica AG) or equivalent (e.g., home-made) freeze-substitution apparatus
UV lamp for polymerization with 350-nm UV light (if not built into the freeze-substitution apparatus)
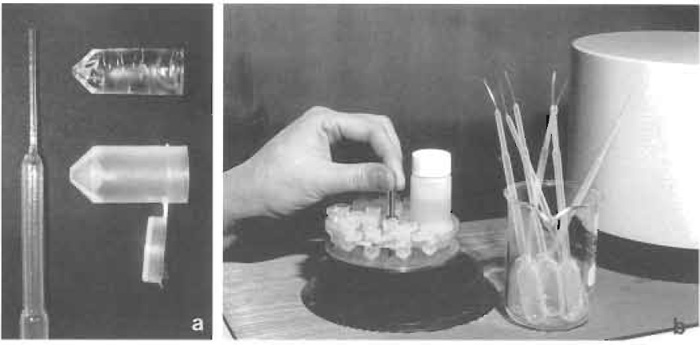 |
| FIGURE 1 Freeze substitution in HM20. (a) Polyethylene Pasteur pipette with elongated tip (left), polyethylene capsule with hinged lid (lower right), and polymerized Lowicryl block after removal of capsule (upper right). (b) Holder with polyethylene capsules and vial for substitution fluid before being lowered into the Balzers FSU 010 freeze-substitution apparatus. To the right beaker with polyethylene Pasteur pipettes and to the far right part of a UV polymerization unit. |
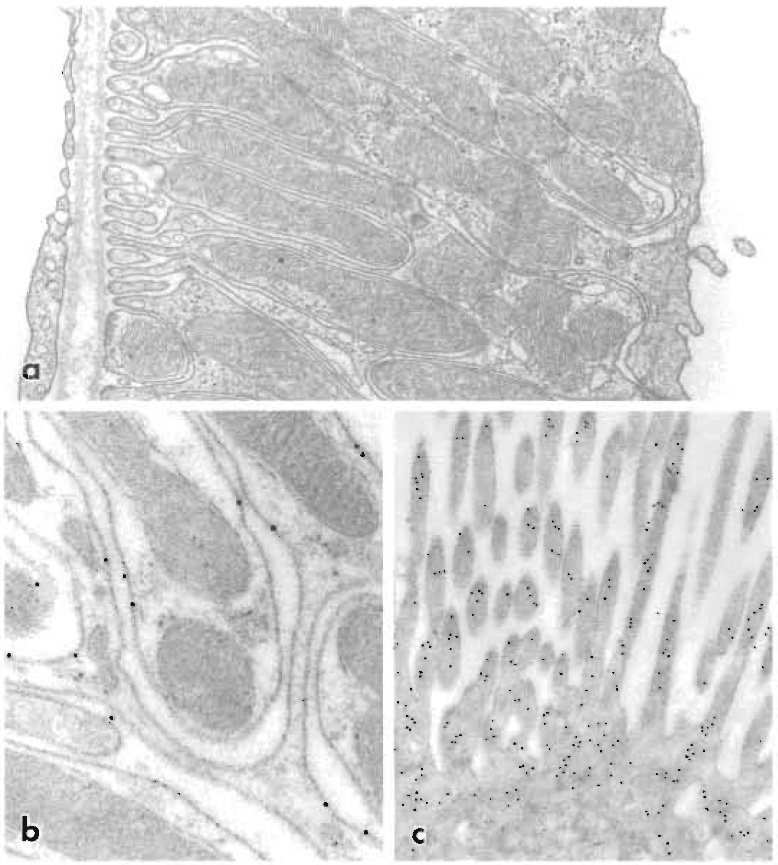 |
| FIGURE 2 Electron micrographs of kidney cells following freeze substitution and embedding in Lowicryl HM20. (a) Thick ascending limb of the renal distal tubule perfusion fixed with 4% paraformaldehyde containing 0.4% picric acid. Cell fine structure is well preserved and cellular membranes stand out in good contrast. Magnification 30,000x. (b) Immunolocalization of Na,K-ATPase to basolateral membranes of distal tubule in rat kidney cortex. Perfusion fixation with 4% paraformaldehyde. The monoclonal antibody against the α subunit of Na,K-ATPase was detected with goat antimouse IgG on 10nm colloidal gold. Note the precise association between basolateral membranes and gold particles. Magnification 80,000x. From Maunsbach (1992). (c) Apical part of proximal tubule cell in kidney perfusion fixed with 2% paraformaldehyde. Intracellular actin was immunolabeled with rabbit antiactin antibody, which was then detected with goat antirabbit IgG on 10nm colloidal gold. Magnification 40,000x. |
B. Procedure for Freeze Substitution in Lowicryl HM20
Freeze substitution and Lowicryl HM20 embedding combine good preservation of cell ultrastructure and antigenicity (Figs. 2 and 4). For immunoelectron microscopy the tissue is usually fixed for short time with 4 or 8% paraformaldehyde or 4% paraformaldehyde plus 0.1% glutaraldehyde but is never postfixed in osmium tetroxide. The tissue blocks should not exceed 1.0mm in any direction.
Solutions
Lowicryl HM20: To make about 20g, gently mix 3.0g cross-linker D and 17.0g monomer E. Bubble dry nitrogen gas into the mixture for 5 min to exclude O2, which inhibits Lowicryl polymerization. Add 0.1 g initiator C and mix gently until it is dissolved. Avoid making air bubbles.
Methanol: Dry the methanol with silica gel.
Methanol: Lowicryl HM20 mixtures in proportions 2:1 and 1:1
2.3M sucrose in phosphate-buffered saline (PBS): To make 100ml solution, weigh out 0.038 g sodium dihydrogen phosphate monohydrate, 0.128g disodium hydrogen phosphate dihydrate, 0.877g sodium chloride, and 78.7g sucrose. Add water to about 95ml. Use a magnetic stirrer until the sucrose is dissolved, or heat the solution in a microwave oven. Adjust pH if necessary to pH 7.2 and fill up with water to 100 ml.
0.5% uranyl acetate in methanol: To make 100ml, dissolve 0.5 g uranyl acetate dihydrate in methanol to a total of 100ml
Steps
- Transfer the tissue directly from the aldehyde fixative to the buffered sucrose solution. Place the specimens in the sucrose for 2h or overnight. Stir the solution occasionally to improve infiltration.
- Cryofix the tissue in liquid nitrogen. Hold the specimen gently with the fine forceps provided with the cold-insulated handle. Remove excess sucrose solution. Move the forceps quickly into the liquid nitrogen and shake until bubbling stops. The specimen is either left temporarily for immediate processing or is placed in a tube for further storage in liquid nitrogen for weeks or months. Carefully follow the safety regulations when handling liquid nitrogen.
- Transfer the frozen tissue very rapidly with a precooled forceps from the liquid nitrogen to the 0.5% uranyl acetate/methanol solution, which is kept at -85 to 90°C in the capsules in the freeze-substitution unit. During this transfer great care must be taken not to warm the specimen. For this purpose the vessel with the liquid nitrogen must be placed immediately adjacent to the substitution unit.
- Withdraw most of the substitution fluid from the capsules with a polyethylene Pasteur pipette. The diameter of the tip of the Pasteur pipette should be smaller than the size of the specimens, as the specimens are difficult to observe at this step and may otherwise be removed. Fill the capsules with temperature-equilibrated substitution fluid of the same composition as before and increase the temperature to -80°C for 24 h. Adjust the following rinsing and infiltration periods to suit regular working hours.
- Rinse the specimens with one change of methanol at -80°C for about 20 h.
- Rinse the specimens with one change of methanol at -70°C for 8 h.
- Rinse the specimens three times with methanol at -45°C over a period of 20 h.
- Infiltrate the specimens with a 2:1 mixture of methanol and Lowicryl HM20 at -45°C for 6h.
- Infiltrate the specimens with an 1:1 mixture of methanol and Lowicryl HM20 at -45°C for about 4h.
- Infiltrate the specimens with pure Lowicryl HM20 at -45°C for 8 h with three changes.
- Infiltrate the specimens with pure Lowicryl HM20 at -45°C for 24h.
- Fill up the capsules completely with fresh Lowicryl HM20 and close the lids. Polymerize with indirect UV light at -45°C for 48 h.
- Increase the temperature to 0°C and continue UV polymerization for 48 h. The specimens are now ready for conventional ultramicrotomy at room temperature.
C. Comments
- Chemicals used during dehydration and embedding resins are toxic (mutagenic, allergenic, and, in some cases, perhaps carcinogenic) and should be handled with adequate safety precautions (Ringo et al., 1982). Work in a well-ventilated hood and use gloves. Note that resins can penetrate most types of gloves within a short time.
- Cryosectioning and freeze substitution followed by Lowicryl embedding are alternative procedures for immunoelectron microscopy. Immunolabeling of sections from freeze-substituted and Lowicryl embedded tissues usually provides a very high precision in probe localization (Figs. 2b, 2c, and 4). A major advantage is that Lowicryl blocks are stable and can be stored at 4 or -20°C and repeatedly resectioned and immunolabeled.
D. Pitfalls
- It is necessary that the specimen is frozen C cryofixed") without ice crystal formation, which will leave holes in the tissue. Tissue blocks should therefore be small and completely infiltrated with 2.3M sucrose before freezing.
- In order to avoid recrystallization of the tissue water during freeze substitution it is important that the temperature of the specimen does not increase when it is tranferred to the freeze-substitution unit or in connection with fluid changes. Fluids must be properly temperature equilibrated in the freezesubstitution unit before being added to the samples.
- The size of the specimen for freeze substitution must be small to allow complete removal of water and of methanol. Remaining water or methanol will result in soft Lowicryl or holes in the tissues. Substitution times may require adjustment according to the tissue studied.
- Capsules should be completely filled with Lowicryl and the lids closed to exclude oxygen during polymerization. Oxygen interferes with polymerization, which results in soft blocks.
III. IMMUNOLABELING OF LOWICRYL SECTIONS
A. Materials and Instrumentation
Disodium hydrogen phosphate dihydrate (Na2HPO4·2H2O, Merck 106580)
Sodium dihydrogen phosphate monohydrate (NaH2PO4·H2O, Merck Cat. No. 1.06346)
Glycine (C2H5NO2, Sigma G-7126)
Bovine serum albumin, BSA (fraction V, Sigma Cat. No. A 4503)
Skimmed milk powder (Merck 115363)
Gelatin (from cold water fish skin, Sigma Cat. No. G7765)
Polyethylenglycol 20,000
Goat antirabbit IgG (or other appropriate antibodies) conjugated to 10-nm (or, e.g., 5nm) colloidal gold particles
Protein A conjugated to 10-nm colloidal gold particles (alternatively)
Uranyl acetate dihydrate [(CH3COO)2UO2·2H2O, Polysciences 21477]
Trisodium citrate dihydrate [(C6H5Na3O7 ·2H2O), Merck 106448]
Lead(II) nitrate [Pb (NO3)2, Merck 107398]
Sodium hydroxide (NaOH, Merck 106498)
Sodium azide (NaN3, Merck 106688)
Sodium chloride (NaCl, Merck 106404)
Redistilled water
Parafilm (or dental wax)
Nickel grids (necessary for immunolabeling)
Fine forceps, antimagnetic
Disposable Pasteur pipettes
Filter paper, 0.2 µm
Magnetic stirrer
B. Procedures
Ultrathin sections of aldehyde fixed and Lowicryl HM20 or K4M-embedded tissue can be used reproducibly and conveniently for immunocytochemical detection of many antigens. Depending on the sensitivity of the antigen, the tissue is fixed either with formaldehyde or mixtures of formaldehyde and glutaraldehyde (see article by Maunsbach). The Lowicrylembedded tissue can be stored, preferably in the cold room, and resectioned repeatedly.
C. Solutions
- Rinsing solution: phosphate-buffered saline (0.01M sodium phosphate buffer containing 0.15M sodium chloride) with 0.1% skimmed milk (or 1% BSA). Mix 0.7ml 0.2M NaH2PO4·H2O and 1.8ml 0.2M Na2HPO4·2H2O, 0.438g NaCl. Adjust pH to 7.4 and fill up with redestilled H2O to make 50ml. Add 0.05 g milk powder (or 0.5 g BSA).
- Preincubation solution: Rinsing solution with 0.05M glycine. Add 0.375g glycine per 100ml rinsing solution.
- Solution for dilution of primary antibody: Same as rinsing solution if skimmed milk is used. If BSA is used, the concentration should be 0.1%.
- Solution for dilution of gold-conjugated antibodies (or protein A): Same as solution for dilution of primary antibody but with the addition of 1.5ml of 1% polyethyleneglycol and 0.555g fish gelatine per 25ml solution (to reduce aggregation of gold particles). The stability of solutions for preincubation, rinsing, and dilution of antibodies can be improved by adding 0.02M sodium azide (NaN3).
 |
| FIGURE 3 Immunolabeling of five grids on series of drops containing preincubation solution, rinsing solution, solution with primary antibodies (AB), rinsing solution, secondary antibody, and distilled water. The grids are transferred between the drops with a forceps. In practice, all drops are not placed at the Parafilm from the very beginning but are applied shortly before use. |
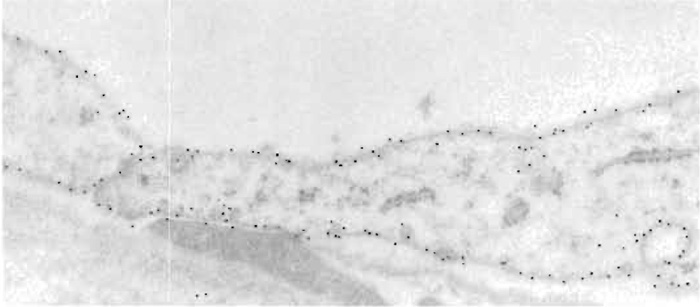 |
| FIGURE 4 Cells in descending thin limb of inner medulla of human kidney labeled with polyclonal antibody against aquaporin-1 water channel. The tissue was fixed in paraformaldehyde and freeze substituted and embedded in Lowicryl HM20. The primary antibody was visualized with goat antirabbit IgG conjugated to 10-nm colloidal gold particles. Cells exhibit strong labeling of both apical and basal plasma membranes. Magnification 60,000×. From Maunsbach et al. (1997) with permission. |
D. Steps
- Place nickel grids with ultrathin sections of Lowricryl section side down on small drops (each 10-25 µl of the appropriate solutions). Place the drops on a piece of Parafilm (Fig. 3), and transfer the grids successively between drops. At each transfer most of the previous solution is removed by quickly touching the grid to a filter paper, but without allowing the grid to dry. Transfer the grid between the following drops.
- Preincubation solution for 15min at room temperature.
- Solution of primary antibody diluted 1:50- 1:5000 depending on the characteristics of antigen and antibody. Incubate for 1 h at room temperature or at 4°C overnight in a moist chamber.
- Rinsing solution three times, 5 min on each drop.
- Secondary antibody (or protein A) conjugated to colloidal gold. Dilution 1:50-1:200, incubation time 1h at room temperature.
- Rinsing solution three times, 5 min on each drop.
- Rinsing water (redestilled and filtered) 5 min on each of three drops.
- Remove excess water with filter paper and air dry the grid.
- Stain for 10min on saturated and filtered uranyl acetate (see Section IV).
- Rinse with 15-20 drops of redestilled water.
- Dry the grid, which is now ready for examination in the electron microscope. Additional contrast can be obtained with lead citrate staining (see Section V).
E. Comments
Electron microscope immunocytochemistry requires careful control of the experimental conditions. As initial steps make sure that the antigen is present in the tissue by immunoblotting and that the antibody specifically and selectively recognizes the appropriate antigen. It is also advisable to apply immunoflourescence and/or immunoperoxidase for overall localization of the antigen. Initial controls at the electron microscope level should include replacement of the primary antibody with buffer solution, with nonimmune serum, and, if possible, with preimmune serum from the same animal that produced the antibodies. Also, if at all available, the antibody should be absorbed with purified antigen before use (usually preabsorbed overnight in the cold room). Some common pitfalls or problems in electron microscope immunocytochemistry are listed next.
F. Pitfalls
- Abscence of labeling or weak labeling may be due to destruction of antigen during fixation and/or embedding. Suggested remedies include (a) decrease concentration of fixative, (b) use formaldehyde alone instead of glutaraldehyde or glutaraldehyde/formaldehyde, (c) shorten fixation time, (d) use another type of Lowicryl, e.g., K4M, (e) use smaller gold particles (e.g., 5 nm; see Giberson and Demaree, 1994), (f) utilize different etching procedures (see Matsubara et al., 1996; Maunsbach and Afzelius, 1998), (g) reduce the salt concentration in the solutions used for dilution of antibodies, and (h) turn to the alternative method of cryoultramicrotomy and labeling of cryosections. Make sure by Western blotting that the antigen exists in the tissue.
- Abscence of labeling or weak labeling may also be due to no or low affinity of the antibody to the antigen, poor titer of the antibody, or the antibody is aged, has been stored improperly, or frozen and thawed repeatedly.
- High backgound labeling may be due to insufficient rinsing of the grid during labeling or too high titer of the primary antibody or secondary antibody (or protein A).
- High background staining may, in some cases, be prevented by increasing the sodium chloride concentration or the pH in the incubation solution for the primary antibody and/or the secondary gold conjugate. If the tissue contains high concentrations of free aldehyde groups following glutaraldeyde fixation, the sections should be exposed to 0.5mM ammonium chloride before incubation.
- Clustered colloidal gold particles may be observed if the primary antibody has aggregated or if the colloidal gold conjugate has aggregated and needs to be renewed.
IV. SECTION STAINING WITH URANYL ACETATE
Uranyl acetate staining (Watson, 1958) results in a uniform but fairly weak general staining of cellular components. There is a slight preference for increased contrast of DNA and RNA.
A. Materials
Uranyl acetate dihydrate [(CH3COO)2UO2·2H2O, Polysciences 21477]
Trisodium citrate dihydrate [(C6H5Na3O7 ·2H2O), Merck 106448]
Lead(II) nitrate [Pb (NO3)2, Merck 107398]
Sodium hydroxide (NaOH, Merck 106498)
Redistilled water
Parafilm (or dental wax)
B. Solution
Saturated solution of uranyl acetate: To make 100ml, dissolve 7.69g of uranyl acetate in 100ml distilled water and stir for some hours. Store the solution at room temperature in the dark in a closed glass vial covered by aluminium foil.
C. Steps
- Filter a suitable volume of stain solution through a 0.2-µm filter immediately before use.
- Place a series of drops on the Parafilm with a clean Pasteur pipette. The number of drops should equal the number of grids to be stained (Fig. 5a).
- Place the grids section side down on the drops with an interval of i min between each grid.
- Leave the grids on the stain drops for 10min.
- Remove and rinse the grids in the same order they were placed on the staining solution. Lift the grid with a clean forceps and hold vertically. Jet slowly a stream of 15-20 drops of filtered water from a Pasteur pipette onto the grid (Fig. 5b).
- Remove the last drop of water by touching the edge of the grid with a filter paper. Place a small piece of filter paper between the legs of the forceps to remove additional excess water.
- Let the grid dry for 5 min while still clamped in the forceps.
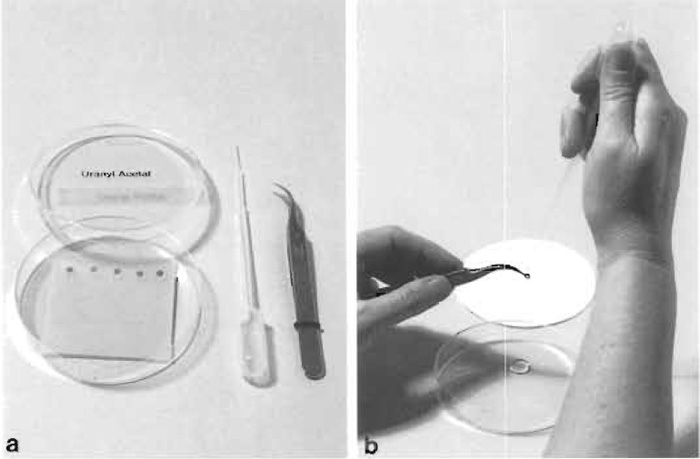 |
| FIGURE 5 (a) Equipment for section staining with uranyl acetate (or lead citrate). With a clean Pasteur pipette, stain drops are placed on dental wax in a petri dish. (b) Rinsing of a stained grid with a stream of drops of redistilled water. Immediately afterward the edge of the grid is touched to filter paper to remove excess water. |
V. SECTION STAINING WITH LEAD CITRATE
Lead staining (Reynolds, 1963) is used when a general survey of tissue ultrastructure is aimed at and gives a general increase in contrast of membranes and other tissue components.
A. Materials and Instrumentation
Trisodium citrate dihydrate [(C6H5Na3O7 ·2H2O), Merck 106448]
Lead (II) nitrate [Pb (NO3)2, Merck 107398]
Sodium hydroxide (NaOH, Merck 106498)
Redistilled water
Parafilm (or dental wax)
B. Solution
Lead citrate solution: Boil about 50ml redistilled water for 5 min in a beaker. Dissolve 1.33 g lead nitrate and 1.76g sodium citrate in 40ml of the boiled water. Stir the solution rapidly for 30min with a magnetic stirrer or shake vigorously. The solution should become clear by adding 8.0ml 1N sodium hydroxide freshly prepared with redistilled water. Fill up to 50 ml with water. The solution should stand for 2 or 3 days before use. Store the solution at 4°C and use within 6-12 months.
C. Steps
- Before use, filter 10ml stain solution through filter paper into a 10-ml cylinder. Close the cylinder with a piece of Parafilm. Handle the cylinder carefully without shaking.
- Take out the stain solution with a clean Pasteur pipette. Remove the solution from the middle of the cylinder to avoid precipitate.
- To stain four grids, place four drops of stain on Parafilm. Do not stain more than four grids at a time, as a precipitate forms easily on the surface of the drops. (Surface precipitate can be minimized by placing NaOH tablets around the grids, but in practice we do not find this necessary.)
- Immediately place the grids on the drops with an interval of half a minute.
- Stain the sections for 2min (range: 15s to 20min depending on desired contrast).
- Remove the grids with intervals of half a minute with fine forceps. Hold the forceps with the grid vertically and squeeze gently from a Pasteur pipette 10-15 drops of redistilled water on the grid (Fig. 5b).
- Touch the grid with filter paper and leave the grid to dry in the forceps with a small piece of filter paper between the legs of the forceps.
D. Pitfalls
- Dirt may contaminate the sections unless all glassware, Parafilm, and pipettes are meticulously clean and all solutions filtered.
- Excess rinsing of lead stain may gradually remove stain and may, in addition, make the staining more susceptible to beam damage in the electron microscope.
- The surface of lead citrate drops rapidly acquires surface contamination due to interaction with carbon dioxide in the air. This will appear as contamination on the section. Therefore, drops must be placed on the Parafilm immediately before the grids are applied to surface of the drops.
E. Comments
The chemicals used for section staining are toxic and should be handled with adequate safety precautions. Staining should be performed in a well-ventilated hood, using gloves. The results of section staining depend in part on the preceding fixation of the tissue and on the characteristics of the embedding medium as well as the protocol of the staining process. Thus if uranyl acetete staining is carried out at 60°C instead of room temperature, contrast is greatly enhanced, which may reveal new tissue components but overstain others. Various instruments and procedures have been described for simultaneous staining of many grids, e.g., a full grid box. The author has never found these procedures helpful.
References
Carlemalm, E., Garavito, R. M., and Villiger, W. (1982). Resin development for electron microscopy and an analysis of embedding at low temperature. J. Microsc 126, 123-143.
Carlemalm, E., Villiger, W., Hobot, J. A., Acetarin J.-D., and Kellenberger, E. (1985). Low temperature embedding with Lowicryl resins: Two new formulations and some applications. J. Microsc. 140, 55-63.
Giberson, R. T., and Demaree, R. S., Jr. (1994). The influence of immunogold particle size on labeling density. Microsc. Res. Tech. 27, 355-357.
Griffiths, G. (1993). "Fine Structure Immunocytochemistry." Springer-Verlag, Berlin.
Larsson, L-I. (1988). "Immunocytochemistry: Theory and Practice." CRC Press, Boca Raton, FL.
Matsubara, A., Laake, J. H., Davanger, S., Usami, S., and Ottersen, O. P. (1996). Organization of AMPA receptor subunits at a glutamate synapse: A quantitative immunogold analysis of hair cell synapses in the rat organ of corti. J. Neurosci. 16, 4457-4467.
Maunsbach, A. B. (1992). Trends in tissue preparation for electron microscopy. In "Electron Microscopy 92" (A. Rfos, J. M. Arias, L. Megias-Megfas, and A. López-Galindo, eds.), Vol. 1, pp. 3-8. University of Granada, Servicio de Publicaciones, Granada.
Maunsbach, A. B., and Afzelius, B. A. (1998). "Biomedical Electron Microscopy: Illustrated Methods and Interpretations." Academic Press, San Diego.
Maunsbach, A. B., Marples, D., Chin, E. Ning, G. Bondy, C., Agre, P., and Nielsen, S. (1997). Aquaporin-1 water channel expression in human kidney. J. Am. Soc. Nephrol. 8, 1-14.
Newman, G. R., and Hobot, J. A. (1993). "Resin Microscopy and On-Section Immunocytochemistry." Springer-Verlag, Berlin.
Reynolds, E. S. (1963). The use of lead citrate at high pH as an electron-opaque stain in electron microscopy.
J. Cell Biol. 17, 208-212.
Ringo, D. L., Brennan, E. F., and Cota-Robles, E. H. (1982). Epoxy resins are mutagenic: Implications for electron microscopists. J. Ultrastruct. Res. 80, 280-287.
Roth, J. (1986). Post-embedding cytochemistry with gold-labelled reagents: A review. J. Microsc. (Oxford) 143, 125-137.
Schwarz, H., and Humbel, B. M. (1989). Influence of fixatives and embedding media on immunolabelling of freeze-substituted cells. Scan. Microsc. Suppl. 3, 57-64.
Sitte, H., Neumann, K., and Edelmann, L. (1989). Cryosectioning according to Tokuyasu vs. rapid-freezing, freeze-substitution and resin embedding. In "Immuno-gold Labeling in Cell Biology" (A. J. Verkleij and J. L. M. Leunissen, eds.), pp. 63-93. CRC Press, Boca Raton, FL.
Watson, M. L. (1958). Staining of tissue sections for electron microscopy with heavy metals. J. Biophys. Biochem. Cytol. 4, 475-478.
Support our developers




