Analysis of Nuclear Protein Import and Export in Digitonin-Permeabilized Cells
Transport between the nucleus and the cytoplasm is mediated by nuclear pore complexes (NPCs), specialized channels that are embedded in the nuclear envelope membrane. Proteins that undergo nuclear import or nuclear export usually encode a nuclear localization signal (NLS) or a nuclear export signal (NES). These signals are recognized by import or export receptors that, in turn, facilitate targeting of the protein to the NPC and translocation through the central channel of the NPC. Both import and export pathways are regulated by the Ras-related GTPase Ran. Nuclear Ran in its GTP-bound form promotes the release of NLSproteins from import receptors after the NLSprotein/ import receptor has reached the nuclear side of the NPC. In contrast, nuclear Ran in its GTP-bound form promotes the assembly of NES-containing proteins with export receptors by forming an NESprotein/ export receptor/RanGTP. Export complexes are then disassembled on the cytoplasmic side of the NPC through the action of a Ran GTPase-activating protein that stimulates GTP hydrolysis by Ran. More specific information regarding import and export complex assembly, models for translocation, and additional aspects of nuclear transport regulation are described elsewhere (Steggerda and Paschal, 2002; Weis, 2003 and references therein).
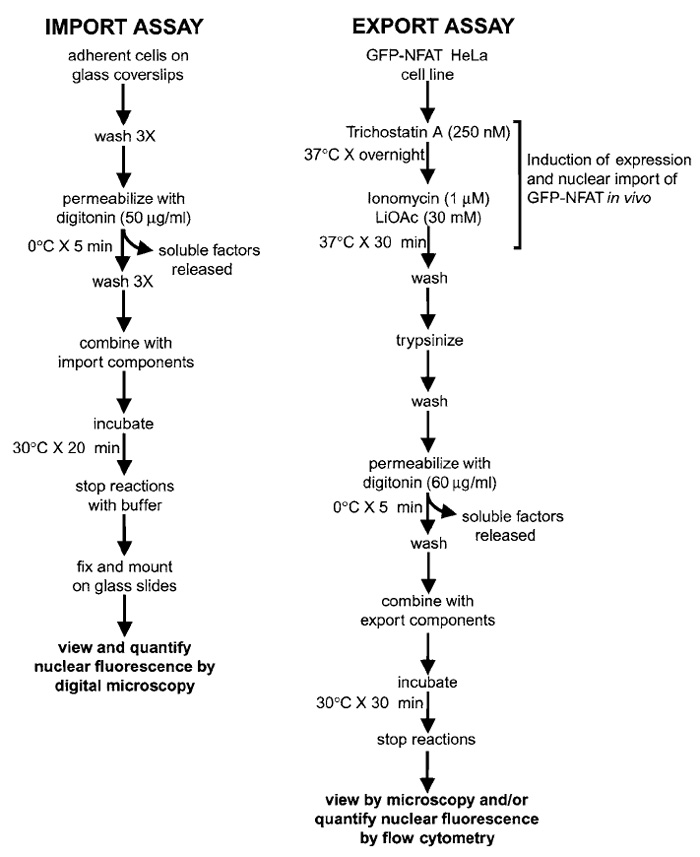 |
| FIGURE 1 Overview of assays for measuring nuclear import and export in digitonin-permeabilized cells. See text for details. |
Rabbit recticulocyte lysate (L4151) is from Promega. HeLa S3 cells (CCL-2.2) and NIH 3T3 cells are from the American Type Culture Collection. Joklik's modified minimum essential medium for suspension cell culture (JMEM; Cat. No. M0518) is from Sigma. Newborn calf serum (Cat. No. 16010-159), penicillin-streptomycin (Cat. No. 15140-122), and trypsin EDTA (Cat. No. 25200-056) are from GIBCO/Invitrogen. ATP (Cat. No. A-2383), creatine phosphate (Cat. No. P-7936), creatine phosphokinase (Cat. No. C-7886), sodium bicarbonate (Cat. No. S- 4019), HEPES (Cat. No. H-7523), potassium acetate (Cat. No. P-5708), magnesium acetate (Cat. No. M- 2545), EGTA (Cat. No. E-4378), dithiothreitol (DTT, Cat. No. D-5545), NaCl (Cat. No. S-9763), potassium phosphate, monobasic (Cat. No. P-0662), phenyl-methylsulfonyl fluoride (PMSF, Cat. No. P-7626), dimethyl sulfoxide (DMSO, Cat. No. D-8779), sodium carbonate (Cat. No. S-7795), trypan blue (Cat. No. T-8154), ionomycin (Cat. No. 1-0634), LiOAc (Cat. No. L-4158), Hoechst 33258 (Cat. No. B-2883), 4',6-diamidino-2-phenyindole, dilactate (DAPI; Cat. No. 9564), and trichostatin A (Cat. No. T-8552) are from Sigma. High-purity bovine serum albumin (BSA, Cat. No. 238031), aprotinin (Cat. No. 236624), leupeptin (Cat. No. 1017101), and pepstatin (Cat. No. 253286) are from Roche. The FITC isomer I (Cat. No. F-1906) and sulfo-SMCC (Cat. No. 22322) are from Molecular Probes and Pierce, respectively. PD-10 columns (Cat. No. 17-0851-01), Cy2 (Cat. No. PA-22000), and Cy5 (Cat. No. PA-25001) are from Amersham Pharmacia Biosciences. Centricon filters (30-kDa cutoff, Cat. No. 4208) are from Amicon. High-purity digitonin (Cat. No. 300410) is from Calbiochem. Vectashield mounting medium (Cat. No. H-1000) is from Vector Laboratories. Six-well dishes (Cat. No. 3516) are from Corning. Glass coverslips (#1 thickness; Cat. No. 12-548A) and the polystyrene tubes used for the transport assays (Cat. No. 2058) are from Fisher. The microscope used for measuring nuclear import in adherent cells is a Nikon E800 equipped with a charge-coupled device camera. The system is linked to a Macintosh computer running OpenLab software for image acquisition. An Olympus IX70 inverted fluorescence microscope is used for analysis of nuclear export. Images from import and export assays are processed using Adobe Photoshop. The flow cytometric analysis system is the FACScan unit from Beckton-Dickinson.
Equipment for centrifugation includes the JS5.2 swinging bucket rotor and J6B centrifuge, the JA-20 fixed angle rotor and J2 centrifuge, and the type 60Ti fixed angle rotor and L7 centrifuge, all from Beckman. Cytosol dialysis is carried out using the collodion vacuum dialysis apparatus (Cat. No. 253310), and 10,000-Da cutoff membranes (Cat. No. 27110) are from Schleicher and Schuell. The homogenizer used for cell disruption is a 0.02-mm-clearance stainless-steel unit (Cat. No. 885310-0015) from Kontes.
III. PROCEDURES
A. Preparation of Cytosol
The rabbit reticulocyte lysate contains all the soluble factors necessary to reconstitute import and export in digitonin-permeabilized cells (Adam et al., 1990). The only preparation involved is dialysis against 1× transport buffer containing 2mM DTT and 1 µg/ml each of aprotinin, leupeptin, and pepstatin (two buffer changes). Reticulocyte lysate is used at 50% (by volume) of transport reactions. This results in a relatively high total protein concentration (~25-40 mg/ml) in transport assays because the reticulocyte lysate contains a high concentration of hemoglobin. A more economical approach, especially if larger quantities of cytosol are needed for protein purification, is to prepare cytosol from suspension culture HeLa cells. In this case, only 1-2mg/ml (final assay concentration) of HeLa cell cytosol is required to obtain a maximum level of protein import or protein export in digitoninpermeabilized cells.
- 10× transport buffer: 200mM HEPES, pH 7.4, 1.1 M potassium acetate, 20mM magnesium acetate, and 5 mM EGTA. To make 1 liter, dissolve 47.6g HEPES, 107.9g potassium acetate, 4.3g magnesium acetate, and 1.9 g EGTA in 800 ml distilled water. Adjust the pH to 7.4 with 10 N NaOH and bring the final volume to 1 liter. Sterile filter and store at 4°C.
- 1× transport buffer: 20mM HEPES, pH 7.4, 110M potassium acetate, 2mM magnesium acetate, and 0.5 mM EGTA. To make 1 liter, add 100ml of 10x transport buffer to 900ml distilled water.
- 1M HEPES stock, pH 7.4: To make 500 ml, dissolve 119.1 g HEPES (free acid) in 400ml distilled water. Adjust the pH to 7.4 with 10 N NaOH and bring the final volume to 500ml. Sterile filter and store at 4°C.
- 1M potassium acetate stock: To make 500ml, dissolve 49 g potassium acetate in 400ml distilled water. Bring the final volume to 500 ml, sterile filter, and store at 4°C.
- 1M magnesium acetate stock: To make 500 ml, dissolve 107.2g magnesium acetate (tetrahydrate) in 400 ml distilled water. Bring the final volume to 500 ml, sterile filter, and store at 4°C.
- 0.2M EGTA stock: To make 500ml, dissolve 38g EGTA (free acid) in 400ml distilled water. Adjust the pH to -7.0 with 10 N NaOH and bring the final volume to 500ml. Store at 4°C.
- Cell lysis buffer: 5mM HEPES, pH 7.4, 10mM potassium acetate, 2mM magnesium acetate, and 1 mM EGTA. To make 500ml, combine 2.5ml 1M HEPES, pH 7.4, 5 ml 1M potassium acetate, 1 ml 1M magnesium acetate, 2.5 ml 0.2M EGTA, and 489ml distilled water. Store at 4°C.
- Phosphate-buffered saline (PBS): To make 1 liter, dissolve 8 g sodium chloride, 0.2 g potassium chloride, 1.44g sodium phosphate (dibasic), and 0.24g potassium phosphate (monobasic) in 900ml distilled water. Adjust the pH to 7.4 and bring the final volume to 1 liter. Store at 4°C.
- Grow HeLa cells at a density of 2-7 × 105 cells per milliliter in a spinner flask (30-50rpm) in a 37°C incubator (CO2 is not required). The medium is JMEM containing 2.0 g sodium bicarbonate and 2.38 g HEPES per liter. Adjust the pH to 7.3, sterile filter, and store at 4°C. Before use, supplement the medium with 10% newborn calf serum and 1% penicillin-streptomycin. The cells should have a doubling time of approximately 18 h, making it necessary to dilute the culture with fresh, prewarmed medium every 1-2 days.
- HeLa cells from a 250-ml culture provide the starting point for scaling up the preparation to 15 liters. This is carried out by sequential dilution of the culture into larger spinner flasks. The culture should not be diluted to a density below 2 × 105 cells/ml. The spinner flasks used for scaling up preparation are 250ml (1 each), 1 liter (1 each), 3 liters (2 each), and 15 liters (1 each). This process generally takes 5 days.
- Perform the cell harvest and subsequent steps at 0-4°C. Collect the cells by centrifugation (300g for 15 min) in 780-ml conical glass bottles in a Beckman J6B refrigerated centrifuge equipped with a JS5.2 swinging bucket rotor. The cell harvest takes about 1 h.
- Wash the cells by sequential resuspension and centrifugation. Two washes are carried out in ice-cold PBS (1 liter each) and one wash is carried out in 1× transport buffer containing 2 mM DTT. The yield from a 15-liter culture should be approximately 40ml of packed cells.
- Resuspend the cell pellet using 1.5 volume of lysis buffer, supplemented with 3µg/ml each aprotinin, leupeptin, pepstatin, 0.5mM PMSF, and 5mM DTT. Allow the cells to swell on ice for 10 min.
- Disrupt the cells by two to three passes in a stainless-steel homogenizer. Monitor the progress of homogenization by trypan blue staining and phasecontrast microscopy. The goal is to obtain -95% cell disruption. Excessive homogenization should be avoided because it results in nuclear fragmentation and the release of nuclear contents into the soluble fraction of the preparation.
- Dilute the homogenate with 0.1 volume of 10x transport buffer and centrifuge in a fixed angle rotor such as the Beckman JA-20 (40,000g for 30min).
- Filter the resulting low-speed supernatant fraction through four layers of cheesecloth. Subject the filtered low-speed supernatant to ultracentrifugation using a fixed angle rotor such as the Beckman type 60 Ti (150,000 g for 60 min).
- Dispense the resulting high-speed supernatant fraction (-50ml, protein concentration-5mg/ml) into 1- and 4-ml aliquots, flash freeze in liquid N2, and store at -80°C indefinitely.
- HeLa cell cytosol is generally subjected to a rapid dialysis step before use in transport reactions. Thaw a 4-ml aliquot of cytosol at 0-4°C and dialyze for 3 h in 1× transport buffer containing 2mM DTT and 1 µg/ml each of aprotinin, leupeptin, and pepstatin (two buffer changes). We use a vacuum apparatus and a collodion membrane to achieve a twofold concentration of the sample (10mg/ml). Dispense the dialyzed, concentrated cytosol into 100-µl aliquots, flash freeze in liquid N2, and store at -80°C.
Synthetic peptides containing an NLS can be used to direct the nuclear import of a variety of fluorescent reporter proteins. FITC- or Cy2-BSA-NLS and Cy5- BSA-NLS are all suitable for measuring import in the fluorescence microscope or in the flow cytometer. Because excitation and emission spectra of Cy5 are distinct from GFP, Cy5-BSA-NLS is ideal for measuring import and GFP-NFAT export in the same cells. Importantly, the average size of the protein conjugate (>70kDa) is too large to allow diffusion through the NPC and it displays low nonspecific binding to the permeabilized cell. Preparation of fluorescent import ligands is carried out in three steps: fluorescent labeling of BSA, modification of the fluorescent BSA with the heterobifunctional cross-linker sulfo-SMCC, and attachment of NLS peptides. Sulfo-SMCC provides a covalent linkage between primary amines on BSA and cysteine present on the N terminus of the NLS peptide.
Solutions
- PBS: To make 1 liter, dissolve 8g sodium chloride, 0.2g potassium chloride, 1.44g sodium phosphate (dibasic), and 0.24g potassium phosphate (monobasic) in 900ml distilled water. Adjust the pH to 7.4 and bring the final volume to 1 liter. Store at 4°C.
- 0.1M sodium carbonate: To prepare 250ml, dissolve 3.1 g sodium carbonate in 200ml distilled water, adjust the pH to 9.0, and bring the final volume to 250ml. Store at 4°C.
- 1.5 M hydroxylamine: To prepare 100 ml, dissolve 10.4 g in a total volume of 100ml distilled water. Store at room temperature.
- 10 mg/ml FITC: Add 1 ml DMSO to 10 mg FITC in an amber vial and vortex to dissolve.
- 20mM sulfo-SMCC: Prepare a 20mM stock of sulfo-SMCC in the following manner. Preweigh a microfuge tube on a fine balance and use a small spatula to add approximately 1-2mg of sulfo-SMCC to the microfuge tube. Reweigh the tube containing the sulfo-SMCC and add DMSO for a final concentration of 8.7mg/ml.
- Dissolve 10mg high-purity BSA in 1 ml sodium carbonate buffer, pH 9.0.
- Stir the BSA solution in a glass test tube with a microstir bar and add 0.1 ml of 10mg/ml FITC. Cover with foil and stir for 60min at room temperature.
- Stop the reaction by adding 0.1ml 1.5M hydroxylamine.
- Separate FITC-labeled BSA from unincorporated FITC by desalting on a PD-10 column equilibrated in PBS, collecting 0.5-ml fractions. The bright yellow FITC-BSA will elute in the void volume of this column.
- Pool the four or five most concentrated fractions, dispense into 1-mg aliquots, and freeze in foilwrapped microfuge tubes at -20°C.
- Combine 1 mg of FITC-BSA with 50 µl of freshly prepared 20 mM sulfo-SMCC and mix end over end for 45 min at room temperature.
- Separate the sulfo-SMCC-activated FITC-BSA from unincorporated sulfo-SMCC by desalting on a PD-10 column equilibrated in PBS. After loading the sample, fill the buffer reservoir of the column with PBS and collect 0.5-ml fractions. The bright yellow FITC-BSA will elute in the void volume as before.
- Pool the three most concentrated fractions of sulfo-SMCC-activated FITC-BSA and combine with 0.3 mg of NLS peptide (CGGGPKKKRKVED). Mix end over end in a foil-wrapped microfuge tube overnight at 4°C.
- Remove unincorporated NLS peptide by subjecting the sample to four cycles of centrifugation and resuspension in 1× transport buffer using a 2-ml 30-kDa cutoff Centricon filter. Follow the manufacturer's recommendations for centrifugation conditions.
- Adjust the FITC-BSA-NLS conjugate to a final concentration of 2 mg/ml, dispense into 50-µl aliquots, flash freeze in liquid N2, and store at -80°C.
The preparation of Cy2- and Cy5-1abeled import substrate is very similar to the method described earlier for the FITC-labeled substrate.
Steps
- Dissolve 2.5mg BSA in 1 ml 0.1M sodium carbonate. Use one vial-activated Cy2 or Cy5 for coupling. Incubate for 40min at room temperature.
- Separate the CyDye-BSA conjugate from the free dye by chromatography on a PD-10 column equilibrated with PBS.
- To activate CyDye-BSA, add sulfo-SMCC to a final concentration of 2mM. Incubate for 30min at room temperature. Remove free cross-linker using a PD-10 column as described earlier.
- Dissolve 1 mg of NLS-peptide (CGGGPKKKRKVED) with activated CyDye-BSA and incubate the solution overnight at 4°C. Remove free peptide and adjust protein concentration as described previously.
- Freeze aliquots in liquid nitrogen and store at -80°C. After thawing, the import substrate can be kept at 4°C in the dark for a few weeks.
Solutions
- 10× and 1 × transport buffer: See Section III,A.
- Complete transport buffer: 1× transport buffer containing 1 µg/ml each aprotinin, leupeptin, pepstatin, and 2 mM DTT.
- 10% digitonin: To make 2ml, add 0.2g highpurity digitonin to 1.7ml DMSO and dissolve by vigorous vortexing, Dispense into 20-µl aliquots and freeze at -20°C.
- 100mM MgATP: To make 5ml, add 0.5ml 1M magnesium acetate and 0.1 ml 1M HEPES, pH 7.4, to 4ml distilled water. Add 275.1mg ATP, dissolve by vortexing, and bring the final volume to 5 ml. Dispense into 20-µl aliquots and freeze at -80°C.
- 250mM creatine phosphate: To make 5ml, add 0.32 g creatine phosphate to 4ml distilled water, dissolve by vortexing, and bring the final volume to 5 ml. Dispense into 20-µl aliquots and freeze at -20°C.
- 2000 U/ml creatine phosphokinase: To make 5ml, dissolve 10,000U creatine phosphokinase in 20mM HEPES, pH 7.4, containing 50% glycerol. Dispense into 1-ml aliquots and store at -20°C.
- Plate NIH 3T3 cells onto glass coverslips in sixwell dishes at a density of ~5 × 104 cells/well and grow overnight.
- Place the six-well dishes on ice. Aspirate media and gently replace with 2 ml ice-cold, complete transport buffer. Aspirate the transport buffer and replace with fresh transport buffer twice, taking care not to disturb the cells. Complete transport buffer in this and subsequent steps refers to ice-cold, 1× transport buffer supplemented with DTT and protease inhibitors.
- To permeabilize the cells, aspirate the transport buffer from each well and immediately add 0.05% digitonin diluted into complete transport buffer. Incubate for 5 min on ice.
- Stop the permeabilization reaction by aspirating the digitonin solution and replacing with complete transport buffer. Wash the cells twice by alternate steps of aspiration and buffer addition.
- Assemble the import reactions in 0.6-ml microfuge tubes on ice. Each reaction contains (final concentration given) unlabeled BSA (5mg/ml), FITCBSA- NLS (25 µg/ml), MgATP (1 mM), MgGTP (1 mM), creatine phosphate (5mM), creatine phosphpkinase (20U/ml), rabbit reticulocyte lysate (25µl), and complete transport buffer in a total volume of 50 µl.
- Create an incubation chamber by lining a fiatbottomed, air-tight box with parafilm and include a moistened paper towel in the chamber as a source of humidity. Place the chamber on ice.
- Using fine forceps, remove each coverslip, wick excess buffer using filter paper, and place cells side up on the parafilm. Pipette the import reaction onto the coverslip surface without introducing bubbles.
- Float the incubation chamber on a 30°C water bath for 20 min.
- Using fine forceps, remove each coverslip, wick most of the import reaction using filter paper, and immediately place back into the wells of the six-well dish.
- Wash the coverslips twice by alternate steps of aspiration and complete transport buffer addition.
- Fix the coverslips by aspirating the complete transport buffer, adding formaldehyde (3.7%) diluted into PBS, and incubating for 15min at room temperature.
- Remove each coverslip, submerge in distilled water briefly, wick excess water, and mount on glass slides using Vectashield. Seal the edges with clear nail polish.
- View the cells by fluorescence microscopy, and quantify the nuclear fluorescence in 50-100 cells per condition using image analysis software such as OpenLab.
Solutions and Reagents
- The HeLa cell line stably expressing GFP-NFAT, which is used for the export assay, has been described in detail (Kehlenbach et al., 1998) and is available upon request.
- 1 mM trichostatin A: Dissolve 1 mg trichostatin A in 3.3ml ethanol. Store in aliquots at -20°C. Trichostatin A is an inhibitor of histone deacetylases and promotes the expression of GFP-NFAT.
- 1mM ionomycin: Dissolve 1 mg ionomycin in 1.34ml DMSO. Store in aliquots at -20°C. Nuclear accumulation of the reporter protein is induced by the calcium ionophore ionomycin.
- Double-stranded oligonucleotides: Dissolve oligonucleotides (5'AGAGGAAAATTTGTTTCATA and 5'TATGAAACAAATTTTCCTCT), each at 200 µM, in 40 mM Tris, pH 7.4, 20 mM MgCl2, and 50 mM NaCl. Anneal by heating to 65°C for 5 min and slow cooling to room temperature. Freeze in aliquots and store at -20°C. The oligonucleotide sequence corresponds to a DNA-binding site of NFAT. It stimulates export of GFP-NFAT about twofold, probably by releasing the protein from chromatin.
- 1× transport buffer with LiOAc: 20mM HEPES, pH 7.4, 80mM potassium acetate, 2mM magnesium acetate, and 0.5mM EGTA. To make 1 liter, dissolve 4.76g HEPES, 7.85g potassium acetate, 3.06g lithium acetate dihydrate, 0.43g magnesium acetate tetrahydrate, and 0.19 g EGTA in 800 ml distilled water. Adjust the pH to 7.4 with 1 N KOH and bring the final volume to 1 liter. Before use, add 1µg/ml each of aprotinin, leupeptin, pepstatin, and 2mM DTT.
- Complete transport buffer: See Section III, D.
- To stimulate expression of GFP-NFAT, add 250nM trichostatin A to stably transfected HeLa cells and incubate overnight. One 15-cm dish containing ~106 cells is sufficient for 30 reactions.
- Induce nuclear import of GFP-NFAT by adding 1 µM ionomycin and 30mM LiOAc directly to the culture media and return the cells to the 37°C incubator for 30min. Lithium inhibits one of the kinases involved in nuclear phosphorylation of NFAT, a step that is required for efficient export in vivo.
- Rinse cells with PBS and remove from dish by adding trypsin EDTA containing 1 µM ionomycin and 30 mM LiOAc. Transfer the cells to 50ml of cold transport buffer with 5% newborn calf serum. Centrifuge for 5 min at 300g at 4°C and wash once in 50ml transport buffer.
- Resuspend cells in complete transport buffer at 107/ml. Add digitonin to 100 µg/ml (1 µl of a 10% stock per 107 cells). Leave on ice for 3 min and check permeabilization with trypan blue. Dilute the cells to 50ml with transport buffer to release soluble transport factors and collect by centrifugation as described previously.
- Preincubation (optional): Resuspend cells in transport buffer containing 30 mM LiOAc at 107/ml and add MgATP (1 mM), creatine phosphate (5mM) and creatine phosphokinase (20 U/ml). Incubate for 15 min in a 30°C water bath. Wash cells with transport buffer. This step results in the depletion of additional transport factors such as CRM1, rendering them rate limiting in the subsequent reaction. The reporter protein largely remains in the nucleus under these conditions.
- Resuspend cells in transport buffer at 3 x 107/ml. Assemble 40-µl transport reactions in FACS tubes: 300,000 permeabilized cells (10 µl), ATP-regenerating system (1mM MgATP, 5mM creatine phosphate, 20U/ml creatine phosphokinase), Cy5-BSA-NLS (25-50µg/ml), l bOVl annealed oligonucleotide, and HeLa cytosol (2mg/ml).
- Incubate tubes in a 30°C water bath for 30min. Control reactions contain the same components but are kept on ice, as nuclear import and export are temperature dependent.
- Stop the reaction by adding 4 ml of cold transport buffer. Centrifuge for 5 min at 400g and 4°C. Remove most of supernatant by aspiration and resuspend cells in residual buffer. Proceed to step 9 or fix cells for microscopic analysis by the addition of 2ml of formaldehyde (3.7% in PBS). Incubate cells for 15min at room temperature, add 1 µl Hoechst 33258 (10 mg/ml in H2O), and incubate for 5 more minutes. Collect cells by centrifugation (300g for 5 min), wash twice with 1 ml PBS, and resuspend the final cell pellet in 15 µl PBS. Apply the cell suspension to a glass slide, cover with a coverslip, and seal with nail polish.
- Measure the fluorescence of 10,000 cells by flow cytometry. In Becton-Dickinson instruments, GFPNFAT (or BSA-NLS, labeled with FITC or Cy2, if only import is analyzed) is detected in FL1 and Cy5-BSANLS in FL4.
- Normalize the mean fluorescence values with respect to a reaction kept on ice.
Several of the basic controls for assaying nuclear import in digitonin-permeabilized cells are shown (Fig. 2). Nuclear import in digitonin-permeabilized cells is stimulated by the addition of HeLa cytosol or reticulocyte lysate, which provides a source of factors, including import receptors and Ran. A low level of cytosol-independent import is usually observed because digitonin permeabilization does not result in the quantitative release of transport factors. Nuclear import is inhibited at low temperature or by incubation with wheat germ agglutinin (0.2-0.5mg/ml), a lectin that binds to NPC proteins and blocks translocation through the pore. The level of nuclear import is quantified by measuring the nuclear fluorescence in 50-100 cells per condition using a commercially available program such as OpenLab or using a public domain program such as ImageJ (http://rsb.info. nih.gov/ij/).
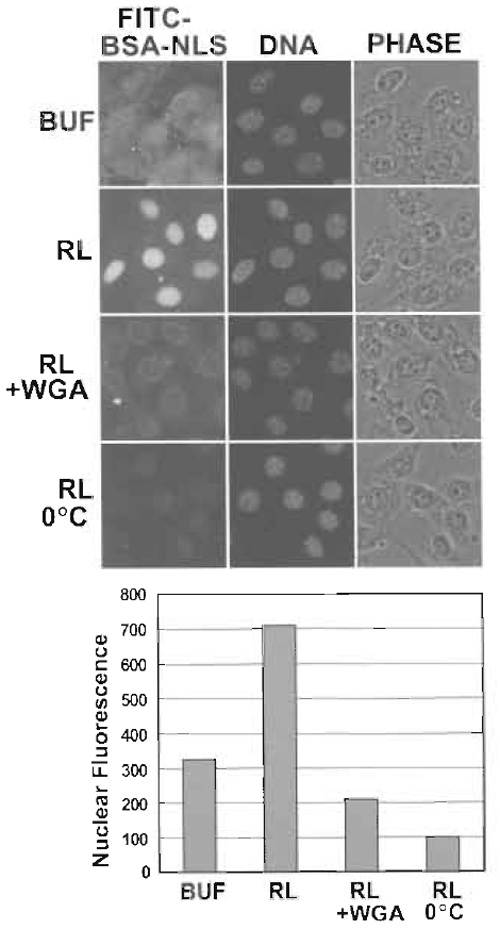 |
| FIGURE 2 Nuclear import of FITC-BSA-NLS analyzed by fluorescence microscopy. (Top) NIH 3T3 cells were digitonin permeabilized and incubated with the indicated components (BUF, buffer; RL, reticulocyte lysate; WGA, wheat germ agglutinin). DAPI staining of DNA and phase-contrast (PHASE) images are also shown. Note that FITC-BSA-NLS binds nonspecifically to cells in the absence of cytosol. The rim fluorescence in the presence of WGA reflects the arrest of FITC-BSA-NLS in import complexes at the NPC. (Bottom) The level of nuclear import observed under each condition was quantified from 12-bit images. Values reflect the average fluorescence intensity per pixel from at least 100 nuclei per condition. Data courtesy of Leonard Shank (University of Virginia). |
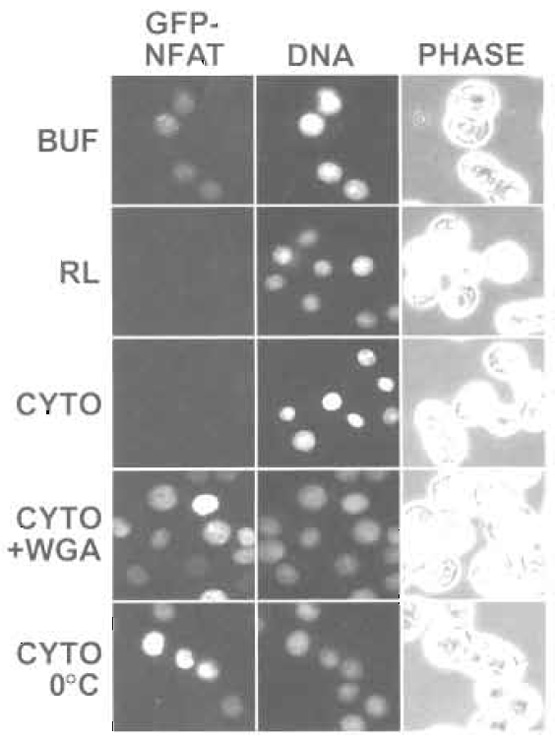 |
| FIGURE 3 Nuclear export of GFP-NFAT was analyzed by fluorescence microscopy. GFP-NFAT cells were treated with trichostatin A and ionomycin to induce expression and nuclear import of GFP-NFAT. After digitonin permeabilization, the cells were incubated with the indicated components (BUF, buffer; RL, reticulocyte lysate; CYTO, HeLa cytosol; WGA, wheat germ agglutinin). Hoechst staining of DNA and phase-contrast (PHASE) images are also shown. |
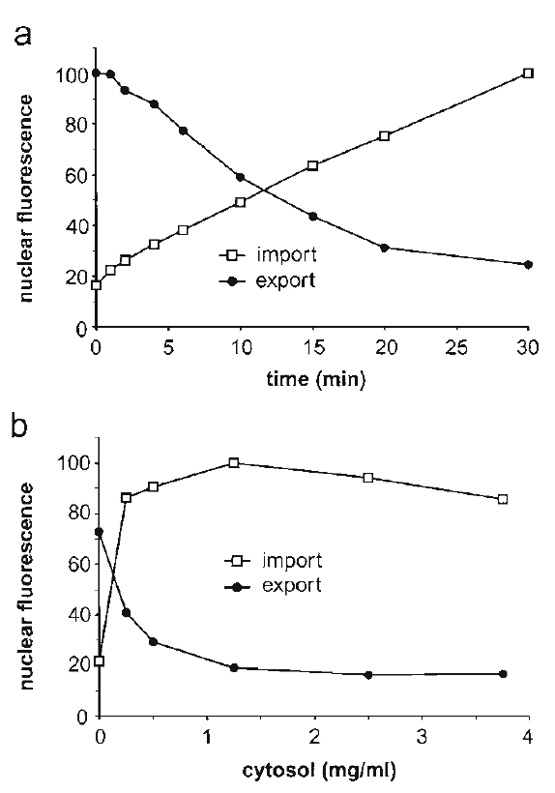 |
| FIGURE 4 Nuclear export of GFP-NFAT and nuclear import of Cy5-BSA-NLS were analyzed in parallel by flow cytometry. (a) Time course of nuclear transport. All reactions contained 2mg/ml of cytosol and 25µg/ml of recombinant Ran. (b) Cytosol dependence of nuclear transport. Reactions contained 50µg/ml of recombinant Ran and the indicated amounts of cytosol. All reactions were performed using preincubated cells (see step 5 in Section III,E) to enhance the cytosol dependence of transport. Portions of this figure were reprinted by permission from the J. Cell Biol., Rockefeller University Press. |
Any compromise in the integrity of the nuclear envelope renders permeabilized cell assays uninterpretable. This could occur if cells are overpermeabilized with digitonin. Under such a condition, NLS-containing reporters can appear to undergo nuclear import when they are, in fact, simply binding to DNA after leakage through a permeabilized nuclear envelope. Likewise, NES-containing reporters can appear to undergo export due to simple leakage from the nucleus. The easiest way to establish that nuclear import or export is mediated by the NPC is to test for inhibition by WGA. Alternatively, the intactness of the nuclear envelope can be demonstrated by showing that a fluorescently labeled dextran (≥70kDa) is excluded from the nucleus. Thus, it is helpful to test a range of digitonin concentrations (25-100µg/ml) and stain with trypan blue to optimize permeabilization of the plasma membrane.
The assays described in this article feature NIH 3T3 and HeLa cells; however, these methods should be applicable to virtually any mammalian cell line. Digitonin permeabilization on adherent cells works best when the cells are 40-70% confluent and poorly if the cells are approaching confluence. Cells that are not well adhered may detach during the permeabilization and wash steps, a problem that usually can be overcome by coating coverslips with poly-D-lysine or by plating the cells 2 days before permeabilization. Also, because cells near the edge of the coverslip may be subject to evaporation artifacts even in a humid chamber, it is best to restrict analysis to the central region of the coverslip.
References
Adam, S. A., Sterne-Marr, R. E., and Gerace, L. (1990). Nuclear import in permeabilized mammalian cells requires soluble factors. J. Cell Biol. 111, 807-816.
Craberee, G. R., and Olson, E. N. (2002). NFAT signaling: Choreographing the social lives of cells. Cell 109, 67-79.
Görlich, D., Pant6, N., Kutay, U., Aebi, U., and Bischoff, E R. (1996). Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 15, 5584-5594.
Görlich, D., Prehn, S., Laskey, R. A., and Hartmann, E. (1994). Isolation of a protein that is essential for the first step of nuclear protein import. Cell 79, 767-778.
Kehlenbach, R. H., Dickmanns, A., and Gerace, L. (1998). Nucleocytoplasmic shuttling factors including Ran and CRM1 mediate nuclear export of NFAT in vitro. J. Cell Biol. 141, 863-874.
Kehlenbach, R. H., and Gerace, L. (2002). Analysis of nuclear protein import and export in vitro using fluorescent cargoes. Methods Mol. Biol. 189, 231-245.
Pollard, V. W., Michael, W. M., Nakielny, S., Siomi, M. C., Wang, E, and Dreyfuss, G. (1996). A novel receptor-mediated nuclear import pathway. Cell 86, 985-994.
Ribbeck, K., and G6rlich, D. (2001). Kinetic analysis of translocation through nuclear pore complexes. EMBO J. 20, 1320-1330.
Steggerda, S. M., and Paschal, B. M. (2002). Regulation of nuclear import and export by the GTPase Ran. Int. Rev. Cytol. 217, 41-91.
Weis, K. (2003). Regulating access to the genome: Nucleocytoplasmic transport throughout the cell cycle. Cell 112, 441-451.
Support our developers




