Heterokaryons: An Assay for Nucleocytoplasmic Shuttling
Interspecies heterokaryon assays are used to analyse the shuttling properties of proteins that localise predominantly in the nucleus at steady state. Conventional localisation methods do not allow the detection of small amounts of protein transiently present in a cellular compartment. Thus, demonstration of a predominantly nuclear (or cytoplasmic) localisation for a given protein frequently obscures the existence of a constant shuttling activity between the two major cell compartments. Indeed, the number of proteins shown to possess nucleocytoplasmic shuttling activity has increased enormously in the past few years, highlighting the importance of shuttling in the regulation of many cellular processes (reviewed by Gama-Carvalho and Carmo-Fonseca, 2001).
Identification of shuttling cytoplasmic proteins can be achieved through analysis of the localisation pattern of deletion mutants affecting export signals or by analysing the effect of inhibition of the major protein export pathway by the drug leptomycin B. In either case, the observation of a shift in the steady-state localisation of the protein from the cytoplasm to the nucleus suggests that the protein under study shuttles continuously between both compartments and that the mutation or drug treatment has interfered with its export pathway.
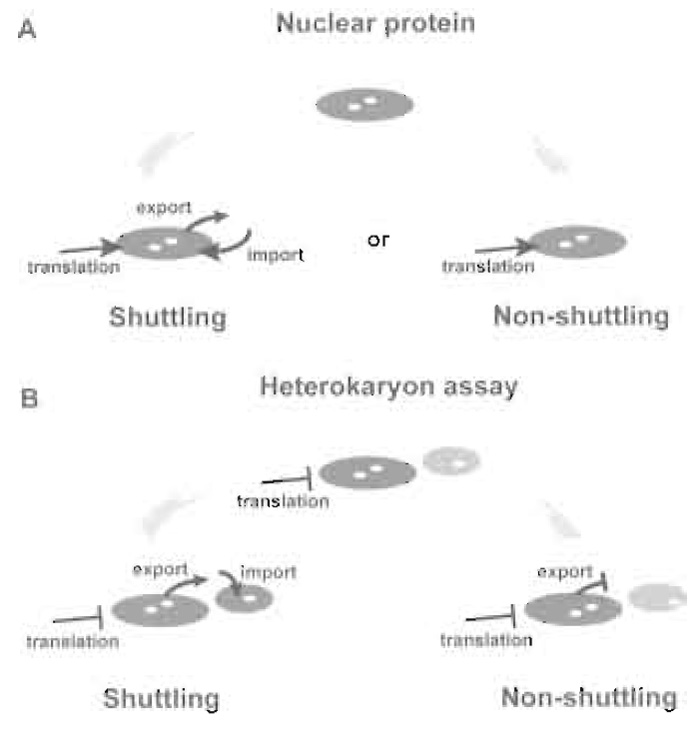 |
| FIGURE 1 The heterokaryon assay. (A) Proteins may accumulate in the nucleus as the result of a unidirectional import pathway (nonshuttling nuclear proteins). Alternatively, proteins may be targeted simultaneously by import and export pathways, resulting in a dynamic cycling (shuttling) between the nucleus and the cytoplasm. (B) Heterokaryon assays distinguish between shuttling and nonshuttling nuclear proteins. If a protein originally present in the donor (large) nucleus shuttles, then it will appear in the receptor (small) nucleus. Nonshuttling proteins are not exported from the donor nucleus and are never detected in the receptor nucleus. Assays have to be performed in the presence of protein synthesis inhibitors in order to prevent the import of newly synthesised protein into the receptor nucleus. |
To perform heterokaryons assays, knowledge of basic tissue culture, transient transfection, and immunofluorescence microscopy techniques is required and will not be addressed here.
Human HeLa cells
Murine NIH 3T3 and/or D. melanogaster SL2 cells Minimum essential medium (MEM) with Glutamax-I (Cat. No. 41090-028) and MEM nonessential amino acids (Cat. No. 11140-035) (Gibco BRL)
Dulbeco's modified Eagle Medium (D-MEM) (Cat. No. 41966-029) or D. melanogaster Schneiders medium (Cat. No. 21720-024) and 200mM L-glutamine (Cat. No. 25030-032) (Gibco BRL)
Fetal bovine serum (FBS, Cat. No. 10270-1064, Gibco BRL)
Basic tissue culture facility with a 37°C, 5% CO2 incubator and a 29°C, 5% CO2 incubator (required only for HeLa × SL2 heterokaryons)
35 × 10-mm (P35) tissue culture dishes and general tissue culture material required for cell line maintenance
10 × 10-mm glass coverslips
Curved tweezer
Transfection reagents: Lipofectin (Cat. No. 18292-037, Invitrogen) or Fugene (Cat. No. 1814443, Roche Applied Science)
Protein synthesis inhibitors: anisomycin (Cat. No. A9789), cycloheximide (Cat. No. L9535) or emetine dihydrochloride hydrate (Cat. No. E 2375) (all from Sigma-Aldrich)
Polyethylene glycol (PEG) 1500 (Cat. No. 783 641, Roche Applied Science)
Primary antibody specific for the human-target protein or protein-fusion construct in eukaryotic expression vector with GFP or amino acid epitope tags (e.g., Ha, FLAG, His) and anti-tag primary antibody
Anti-hnRNP C mAb 4F4 (Choi and Dreyfuss, 1984) and anti-hnRNP A1 9H10mAb (Pinol-Roma et al., 1988)
DAPI, dilactate (Cat. No. D 9564, Sigma-Aldrich) Fluorochrome-conjugated secondary antibodies (Jackson Laboratories)
Fluorescence microscope
III. PROCEDURES
A. HeLa × SL2 Heterokaryons
Cell Culture
- Maintain HeLa cells routinely in MEM supplemented with nonessential amino acids and 10% FBS and grow in a 37°C, 5% CO2 incubator. Cells should be split the day before the heterokaryon assay is performed.
- Grow SL2 D. melanogaster cells in Schneider's medium supplemented with 12% FBS and 2 mM glutamine at 25°C without CO2 (a clean laboratory drawer will provide a convenient "incubator"). This is a suspension cell line that can be induced to adhere to the coverslips in the absence of serum.
- Heterokaryon medium: HeLa × SL2 heterokaryons should be maintained in MEM supplemented with nonessential amino acids and 12% FBS in a 29°C, 5% CO2 incubator.
- Protein synthesis inhibitors: Prepare a stock solution at 10mg/ml in 50% ethanol and store at -20°C. Final use concentrations should be established for the cell lines used; most widely used concentrations range between 20 and 100µg/ml for cicloheximide or anisomycin and 0.5 and 20µg/ml for emetine.
- Phosphate-buffered saline PBS: 137mM NaCl, 2.68mM KCl, 8.06mM Na2HPO4, and 1.47mM KH2PO4. Prepare by weighing 8 g NaCl, 0.2 g KCl, 1.14 g Na2HPO4, and 0.2 g KH2PO4 for 1 liter of solution. Sterilise by autoclaving.
Steps
- All the steps of the procedure should be performed in a laminar flow hood using sterile cell culture material and solutions. The procedure is adapted for cells grown on 10 x 10-mm coverslips placed in P35 dishes.
- HeLa cells should be grown to subconfluent density on 10 x 10-mm coverslips. A 35 x 10-mm tissue culture petri dish can accommodate four of these coverslips.
- Resuspend exponentially growing D. melanogaster SL2 cells in serum-free HeLa culture medium to a concentration of 3-4 × 107 cells/ml.
- Remove the medium from the HeLa cell culture and overlay coverslips with 500µl of the SL2 suspension. Incubate for 20 min in a 29°C, 5% CO2 incubator to induce adherence of SL2 cells.
- Replace medium with 1.5 ml heterokaryon medium and add the protein synthesis inhibitor. Place cells in the 29°C, 5% CO2 incubator for at least 3 h to inhibit protein synthesis. A control experiment replacing the protein synthesis inhibitor with a similar volume of 50% ethanol should be performed.
- For the fusion procedure, place a 50-µl drop of PEG 1500 prewarmed to 29°C in a sterile petri dish.
- Remove the HeLa/SL2 coculture from the incubator and wash twice with PBS prewarmed to 29°C.
- With a sterile forceps, remove a coverslip and invert it over the PEG drop for 2 min (cell side down). Then, place the coverslip (cell side up) in a new P35 dish and rinse twice with PBS prewarmed to 29°C. Replace the PBS with 1.5ml of prewarmed heterokaryon medium with a protein synthesis inhibitor and place the heterokaryons in the incubator for the duration of the shuttling assay (usually 6h or more).
- Fix and immunostain cells using standard procedures, following a time course with 1-h intervals. Positive and negative control experiments should be performed using monoclonal antibodies directed against human hnRNPA 1 (a protein that shuttles between the nucleus and the cytoplasm; Pinol-Roma et al., 1988) and human hnRNPC (a protein that does not shuttle; Choi and Dreyfuss, 1984) (Fig. 2).
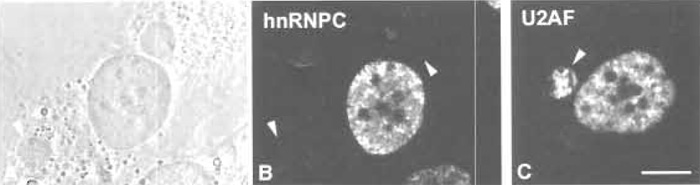 |
| FIGURE 2 HeLa × SL2 heterokaryons. Monoclonal antibodies directed against a human protein often do not cross-react with its Drosophila melanogaster homologue. In this case, shuttling of the endogenous human protein can be analysed in a HeLa × SL2 heterokaryon. (A) Phase-contrast image of a HeLa × SL2 heterokaryon. Arrowheads label small SL2 nuclei from cells that have fused with the HeLa cytoplasm. (B) The heterokaryon shown in A has been immunostained with a monoclonal antibody specific for the human non-shuttling protein hnRNPC (mAb 4F4; Choi and Dreyfuss, 1989), showing it to be restricted to the HeLa nucleus in the presence of protein synthesis inhibitors (20µg/ml emetine). Arrowheads point to the position of SL2 nuclei. (C) Immunostaining of a HeLa × SL2 heterokaryon with a monoclonal antibody specific for human U2AF65 (mAb MC3; Gama-Carvalho et al., 1997) shows that the human protein is present in the SL2 nucleus (arrowhead) in the presence of protein synthesis inhibitors, demonstrating the shuttling activity of this protein (Gama-Carvalho et al., 2001). |
Cell Culture
- Maintain HeLa cells routinely in MEM supplemented with nonessential amino acids and 10% FBS and grow in a 37°C, 5% CO2 incubator. Cells should be split to petri dishes 2 days before the heterokaryon assay is performed and transfected on the following day.
- Grow mouse 3T3 cells in D-MEM supplemented with 10% FBS at 37°C, 5% CO2.
- Heterokaryon medium: HeLa × 3T3 heterokaryons should be maintained as 3T3 cells.
Solutions
- PBS: 137mM NaCl, 2.68mM KCl, 8.06mM Na2HPO4, and 1.47 mM KH2PO4. Prepare by weighing 8 g NaCl, 0.2 g KCl, 1.14 g Na2HPO4, and 0.2 g KH2PO4 for 1 liter of solution. Sterilise by autoclaving.
- DAPI: Prepare a stock solution at 1 mg/ml and store at -20°C protected from light; staining solution: dilute stock to 1 µg/ml in PBS and store at +4°C protected from light for several weeks.
- Protein synthesis inhibitors: Prepare a stock solution at 10mg/ml in 50% ethanol and store at -20°C. Final use concentrations should be established for the cell lines used; most widely used concentrations range between 20 and 100µg/ml for cicloheximide or anisomycin and 0.5 and 20µg/ml for emetine.
- All the steps of the procedure should be performed in a laminar flow hood using sterile cell culture material and solutions. The procedure is adapted for cells grown on 10 x 10mm coverslips placed in P35 dishes.
- The evening before the assay, HeLa cells should be transfected with the desired construct and left to grow to subconfluent density on 10 x 10-mm coverslips. A 35 x 10-mm tissue culture petri dish can accommodate four of these coverslips. Common transfection reagents such as Lipofectin or Fugene work well with this procedure and can be used according to the instructions of the manufacturer. The number of hours between transfection and the heterokaryon assay should be determined for each construct to avoid overexpression of the exogenous protein.
- Tripsinise and count 3T3 cells. For each P35 HeLa culture, prepare 1.5 ml of a 3T3 suspension with 1 × 106 cells/ml in DMEM with 10% FBS and protein synthesis inhibitor.
- Remove the medium from the HeLa cell culture and overlay coverslips with the 3T3 suspension. Incubate for 3 h in a 37°C, 5% CO2 incubator to allow 3T3 cells to adhere and inhibit protein synthesis.
- For the fusion procedure, place a 50-µl drop of prewarmed PEG 2000 in a sterile petri dish per coverslip.
- Remove the HeLa/3T3 coculture from the incubator and wash twice with prewarmed PBS.
- With a sterile forceps, remove a coverslip and invert it over the PEG drop for 2min. Then place the coverslip (cell side up) in a new P35 dish and rinse twice with prewarmed PBS. Replace the PBS with 1.5ml of prewarmed heterokaryon medium with protein synthesis inhibitor and place the heterokaryons in the incubator for the duration of the shuttling assay (usually from 1 to 5 h).
- Fix and stain cells using standard procedures, following a time course with 1-h intervals. In addition to the procedure (if any) necessary for detection of the exogenous protein, cells should be stained with DAPI to allow identification of heyterokaryon nuclei. Positive and negative control experiments should be performed using monoclonal antibodies directed against human hnRNPA 1 (a protein that shuttles between the nucleus and the cytoplasm; Pinol-Roma et al., 1988) and human hnRNPC (a protein that does not shuttle; Choi and Dreyfuss, 1984) (Fig. 3).
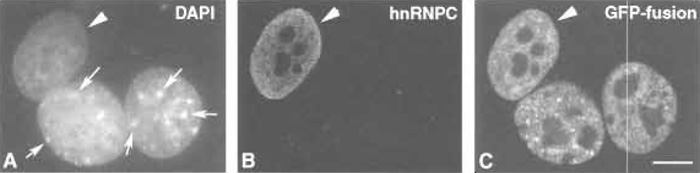 |
| FIGURE 3 HeLa × 3T3 heterokaryons. HeLa cells were transiently transfected with a vector encoding GFP-U2AF65 (Garna-Carvalho et al., 2001) and fused with rnurine NIH 3T3 cells in the presence of 20µg/ml ernetine. A single heterokaryon labelled with DAPI (A), anti-hnRNPC rnAb (B), and GFP (C) is shown. (A) DAPI staining of the HeLa × 3T3 heterokaryon. Murine cells are easily distinguished by the presence of brightly stained blocks or pericentric heterochrornatin (arrows). (B) The heterokaryon shown in A was stained with an antibody specific for the nonshuttling human hnRNP C protein (rnAb 4F4; Choi and Dreyfuss, 1989). hnRNP C is restricted to the HeLa cell nucleus (arrowhead), confirming that there is an efficient inhibition of protein synthesis. (C) Detection of GFP in the HeLa × 3T3 heterokaryon reveals that the fusion protein originally present in HeLa cells shuttles between the nucleus and the cytoplasm. Bar: 10µm. |
Shuttling rates vary significantly from protein to protein, with some being extremely slow (e.g., nucleolin takes up to 16h to be detected in the receptor nuclei) and others very fast (e.g., transport receptors are detected in receptor nuclei within minutes after the fusion). The ideal time period for a shuttling assay should thus be determined for each case by performing a time course analysis. As a general rule, detection of the protein in receptor nuclei in HeLa × 3T3 heterokaryon assays involving transient expression of exogenous protein is significantly faster than HeLa × SL2 assays with the endogenous protein. For example, for the same protein, equilibrium between donor and receptor nuclei may be achieved in 90min in the first case and take up to 7h in the second.
Proper interpretation of results from a heterokaryon assay requires the use of adequate controls. Whenever possible, shuttling activity should be demonstrated for both endogenous and exogenously expressed proteins, as amino acid tags and protein overexpression may modify the results. When assaying for shuttling of the endogenous protein, the specificity of the antibody has to be demonstrated carefully. The efficiency of protein synthesis inhibition must be controlled properly as different cell strains may show a wide variation in the sensitivity to protein synthesis inhibitors. Demonstration of efficient translation inhibition can be performed by staining the heterokaryons with a monoclonal antibody specific for a human nuclear nonshuttling protein, such as the hnRNP C protein (mAb 4F4, Choi and Dreyfuss, 1984). If cytoplasmic protein synthesis is going on, this protein will be detected in the nucleus of the receptor cell. Whenever possible, heterokaryons should be double stained for the protein of interest and hnRNPC. Alternatively, when the protein of interest is also detected with a mAb, a parallel assay should be performed in which the heterokaryons are labelled with the anti-hnRNP C antibody.
The use of interspecies heterokaryons in association with transient transfection opens the possibility to analyse the protein domains involved in the export pathway by the use of mutant forms of the protein. However, this analysis can only be performed with mutants that retain full nuclear localisation. Moreover, leakage of small deletion mutants to the cytoplasm should be monitored carefully.
The confluence of HeLa cells is critical for the procedure. If the density of the culture is too low, few heterokaryons will form. If cells have reached confluence, there will be no space left for the SL2 or 3T3 cells to adhere and often their nuclei will be above HeLa nuclei, making analysis of the results impossible. HeLa coverslips should be checked one by one to choose those with an appropriate subconfluent density.
Often during the fusion procedure there is a significant loss of cells. This may result from the mechanical stress generated when the coverslips that were turned over the PEG droplet are picked. To avoid this, simply pipette PBS beneath the coverslip to make it float and then pick it up with a forceps. In addition, it is possible that the protein synthesis inhibitor is inducing apoptosis of a high percentage of cells in culture. Indeed, some cell strains are highly sensitive to these inhibitors at concentrations that have no negative effects for other cells. Thus, the choice of inhibitor and concentration should be assayed carefully for the particular cell strain used in the assay. Cycloheximide is the most widely used inhibitor, although some cell lines may be more sensitive to emetine. In some cases, efficient protein synthesis inhibition may be hard to achieve, with a significant proportion of cells in the culture showing significant levels of newly translated protein. To prevent this from influencing the interpretation of the assay, perform double labelling of heterokaryons with the anti-hnRNP C 4F4 mAb whenever possible and consider only heterokaryons in which this protein is restricted to the human nuclei.
As discussed earlier, when analysing the shuttling ability of transiently transfected proteins, it is crucial to consider only low expressing cells. A good fluorescence microscope is essential for detecting low levels of expression. Often, multinucleated heterokaryons form. These should not be considered in the shuttling assay, as it is hard to identify the donor nucleus and access the original expression level of the protein (as it has spread out to many nuclei).
References
Almeida, E, Saffrich, R., Ansorge, W., and Carmo-Fonseca, M. (1998). Microinjection of anti-coilin antibodies affects the structure of coiled bodies. J. Cell Biol. 142, 899-912.
Bellini, M., and Gall, J. G. (1999). Coilin shuttles between the nucleus and cytoplasm in Xenopus laevis oocytes. Mol. Biol. Cell 10, 3425-3434.
Calado, A., Kutay, U., Kuhn, U., Wahle, E., and Carmo-Fonseca, M. (2000). Deciphering the cellular pathway for transport of poly(A)-binding protein II. RNA 6, 245-256.
Choi, Y. D., and Dreyfuss, G. (1984). Monoclonal antibody characterization of the C proteins of heterogeneous nuclear ribonucleoprotein complexes in vertebrate cells. J. Cell Biol. 99, 1997-2204.
Gama-Carvalho, M., and Carmo-Fonseca, M. (2001). The rules and roles of nucleocytoplasmic shuttling proteins. FEBS Lett. 498, 157-163.
Gama-Carvalho, M., Carvalho, M. P., Kehlenbach, A., Valcarcel, J., and Carmo-Fonseca, M. (2001). Nucleocytoplasmic shuttling of heterodimeric splicing factor U2AF. J. Biol. Chem. 276, 13104-13112.
Gama-Carvalho, M., Krauss, R. D., Chiang, L., Valcarcel, J., Green, M. R., and Carmo-Fonseca, M. (1997). Targeting of U2AF65 to sites of active splicing in the nucleus. J. Cell Biol. 137, 975-987.
Lee, M. S., Henry, M., and Silver, P. A. (1996). A protein that shuttles between the nucleus and the cytoplasm is an important mediator of RNA export. Genes Dev. 10, 1233-1246.
Leung, A. K., and Lamond, A. I. (2002). In vivo analysis of NHPX reveals a novel nucleolar localization pathway involving a transient accumulation in splicing speckles. J. Cell Biol. 157, 615-629.
Pinol-Roma, S., Choi, Y. D., Matunis, M. J., and Dreyfuss, G. (1988). Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev. 2, 215-227.
Support our developers




