Chromosome Microdissection Using Conventional Methods
Chromosome Microdissection Using Conventional Methods
I. INTRODUCTIONDuring cytogenetic analysis there are frequently complex chromosomal structural aberrations, which are unidentifiable by chromosome banding analysis. Fluorescence in situ hybridization (FISH) combined with G-banding analysis (Xu and Wang, 1994) has significantly improved the accuracy of chromosome identification. However, the efficacy of FISH analysis depends on the probes chosen, which in turn depend on the G-banding result. It is very difficult to choose the adequate probe(s) for FISH analysis when no clue is available from banding analysis. Therefore, FISH can serve as a tool for verification and confirmation. The recently developed multicolor or spectral karyotyping highly facilitates the identification of complex interchromosomal structural abnormalities. However, its application is limited to nonhomologous rearrangements and its efficacy depends on the size of the chromosome segment involved. Furthermore, it can identify the chromosomes involved but not the specific segment or breakpoints involved.
A. Materials
Methyl alcohol (Fisher Scientific, #MD3017-4)
Potassian chloride solution 0.075N (Irvine Scientific, #9281)
Acetic acid, glacial ACS (Fisher Scientific, #A38-500)
Colcemid 10µl/ml in HBSS (GIBCO, #15210-040)
Trypsin (1:250) (GIBCO, #27250-018)
KaryoMax Giemsa stain (Lab Chem Inc., #2C 14840-7)
Hank's balanced salt solution ×1; without CaCl2 + Mg (Sigma, #119349)
Buffer tablets "GURR" (GIBCO, #10582-013)
Deionized distilled (dd)water
T7 DNA polymerase (Sequenase, Version 2.0, USB #70755Y)
Topoisomerase 1 (Promega, #M2851)
Biotin-16-dUTP (Boehringer Mannheim, #1093070)
Taq DNA polymerase (Applied Biosystems, #4311816)
Universal primer 5'-CCGACTCGAGNNNNNNNATGTGG-3' (Telenius et al., 1992)
15-ml sterile polystyrene tubes (Corning, #43005)
Microneedle (Sutter Instrument Co., #BR100-15)
Coverslips (VWR, 24 × 60mm #48393-106)
B. Instrumentation
Incubator, 37°C
Inverted microscope (Nikon DIAPHOT-TMD)
9-in. Pasteur glass pipettes (VCR, #53283-915) and bulbs
Micromanipulators (Narashige, Models MM-88 and MO-302)
Micropipete puller (Narishige, Model PB-7)
Thermal cycler (MJ Research Inc., Model PTC-200)
Slide warmer (VWR)
Coplin jars (0.50-ml capacity)
Thin-walled microtubes (Laboratory Products Sales, #430)
Microcon YM-100 columns (Millipore, #42413)
III. PROCEDURES
A. Preparation of Cells for Microdissection
This procedure is modified from those of Wang and Federoff (1972) and Xu et al. (1994).
- Colcemid rnetaphase arresting solution: 10µg/ml (Sigma D1925)
- Hypotonic solution: Prepare daily by mixing 0.4% KCl (EM Science PX1405-1) (in dd H2O) and 0.4% sodium citrate (EM Science PX0445-1) (in dd H2O) in equal volume
- Fixation solution: Prepare daily by mixing methanol (Mallinckrodt CAS 67-56-1) and glacial acetic acid (Fisher Scientific A38-500 CAS 64-19-7) in a ratio of 3:1 (v/v)
- Trypsin solution: 0.625g trypsin (1:250; GIBCO #27250-018) in 100ml dd H2O
- GURRS buffer solution: Dissolve one GURRS buffer tablet (GIBCO 10582-013) in 1 liter dd H2O, adjust pH to 6.8
- Giemsa staining solution: 2-3ml Giemsa (LabChem Inc. LC14840-7) in 50ml GURRS buffer solution
- Grow cells in cell culture medium supplemented with 10% fetal bovine serum at 37°C in a 6% CO2 humidified incubator.
- Prepare chromosome metaphase spreads from a proliferative cell population using colcemid (5- 10µl/ml) for metaphase arresting, hypotonic treatment followed by fixation for chromosome harvesting.
- Harvest cells either in situ or in suspension and then drop on 24 × 60-mm #1.5 coverslips.
- Perform Giemsa-trypsin-G-banding analysis (GTG) by standard procedure (Wang and Federoff, 1972) for identification of the target chromosome or chromosome segment for microdissection using an inverted microscope.
B. Chromosome Microdissection Combinded with FISH Analysis (Micro-FISH)
The procedure of micro-FISH is performed according to Guan et al. (1993) with modification (Xu et al., 1995).
- 0.625% banding trypsin: Add 0.25g trypsin (1:250) powder to 100ml dd H2O, mix well, aliquot to 2.0ml, and store at -20°C
- GURR buffer: Reconstitute with ddH2O (1 tablet/ 1 liter), adjust pH to 6.8
- Collection buffer (5µl): 40mM Tris-HCl, pH 7.5, 20mM MgCl2, 50mM NaCl, 200µM of each dNTP, 1 unit Topo 1, and 5 pmol of universal primer
- PCR reaction mixture (50µl): 10mM Tris-HCl, pH 8.4, 2mM MgCl2, 50mM KCl, 0.1mg/ml gelatin, 200 µm of each dNTP, 50 pmol universal primer, and 2 units Taq DNA polymerase.
- T7 DNA polymerase working solution: Dilute T7 DNA polymerase (13U/µl) with dilution buffer to 1.5U/µl.
- Labeling solution (50 µl): 100 mM Tris-HCl, pH 8.4, 2mM MgCl2, 50mM dcl, 0.1mg/ml gelatin, 200µl 4dNTP, 50pmol universal primer, 20µM biotin-16- dUTP, and 2 units Taq DNA polymerase.
- Whole chromosome painting probes obtained from commercial laboratories (Vysis)
1. Chromosome Microdissection and DNA Amplification
Steps
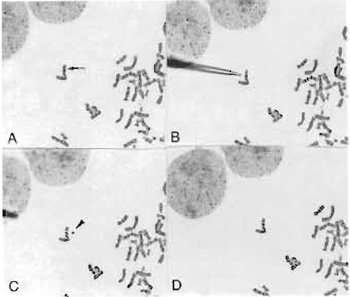 |
| FIGURE 1 Microdissection procedure in progress. The glass needle is aligned using a micromanipulator and the chromosome band or region is dissected. The needle is then used to pick up the dissected piece (C) and transfer it into a collection tube. |
- Prior to microdissection, fix cells in fixative solution by three washes.
- Prepare metaphase spreads on 24 x 60-mm #1.5 coverslips and stain by GTG banding.
- Make microneedles with the micropipette puller according to the manufacturer's manual and UV treat overnight to rule out background DNA contamination.
- Perform microdissection on the target chromosome/ segment, which is identified by its specific G-banding pattern, with glass microneedles (Fig. 1) controlled by a Narashige micromanipulator (Models MM-88 and MO-302) attached to an inverted microscope.
- Transferr the dissected chromosome adherent to the microneedle to a 0.2-ml microcentrifuge tube containing 5 µl of collection buffer.
- Cover the collection buffer with 45 µl of mineral oil and incubate at 37°C for 30min, followed by heat soak at 94°C for 30min.
- Use a gene Amp PCR System 9600 (Perkin-Elmer Atus) for DNA amplification.
- Perform an initial eight cycles of PCR (denaturation at 94°C for lmin, anneling at 30°C for 2min, and extension at 37°C for 2min) by adding approximately 0.3 units of T7 DNA polymerase (Sequence Version 2.0, USB) at each cycle.
- Add 50µl of PCR reaction mixture to the tube.
- Heat the reaction to 95°C for 3 min followed by 35 cycles at 94°C for lmin, 56°C for lmin, 72°C for 2min, with a final extension at 72°C for 5 min.
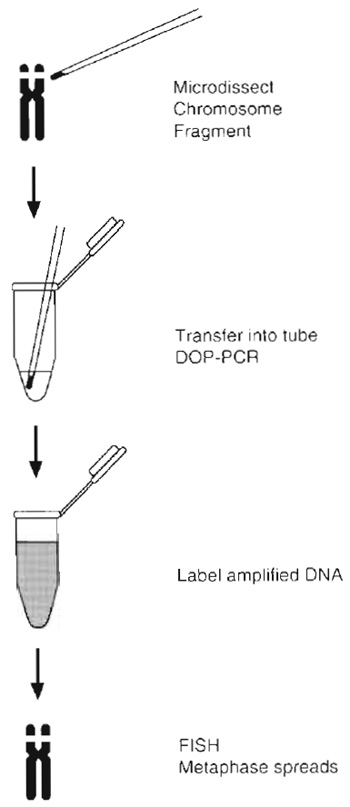 |
| FIGURE 2 Diagram and flowchart of the micro-FISH procedure. Once the whole chromosome or segment is dissected, it is transferred into a collection tube and PCR amplified using random primers. An aliquot of this reaction is then reamplified by PCR with the addition of biotin/digoxigenin dUTP. The labeled PCR products are column purified for use as FISH probes. |
Steps
- Label a 2-µl aliquot of the PCR product for 16 cycles in a 50-µl secondary PCR with the same procedure as described earlier, except for the addition of 20 mM biotin-11-duTP (BMB).
- Purify the labeled PCR products using a Centricon 100 (Amicon) filter.
- Add 100µg of the probe to a 10-µl hybridization mixture containing 55% formamide, 10% detran, 1 x SSC, and 5-10µg COT-1 DNA (BCL).
3. Verification of the Specificity of the Dissected Chromsome/ Segment
Steps
- Grow the cells with the dissected chromosome/ segment on 22 x 22-mm coverslips in 35 x 10-mm petri dishes.
- Block the cells by treatment with colcemid (Gibco), 0.1µg/ml for 1 h, hypotonically treated, fixed, and harvested using standard procedures.
- G-band the chromosomes following trypsin treatment and stain with Giemsa stain.
- After GTG-banding and karyotyping, destain slides with xylene, xylene/ethanol (1/1, v/v), and methanol/acetic acid (3/1, v/v), refix in 3.7% formaldhyde/phosphate-buffered saline (PBS), air dry, and reuse for FISH as described by Klever et al. (1991) using the biotin-labeled probe generated from the dissected chromosome/segment to verify the specificity of the probe.
- Once the specificity of the probe is confirmed, the probe can be hybridized back to normal metaphase spreads to identify the origin of the dissected chromosome segment.
4. Identification of the Origin of the Dissected Chromosome/Segment(s)
- Analyze normal metaphase spreads by GTG banding.
- Apply FISH analysis with the probe specific for the dissected chromosome/segment to normal metaphase spreads after GTG-banding analysis using the procedure described in the previous section.
- Combine the result obtained from GTG banding with that obtained from FISH analysis to identify the origin or components of the chromosome/segments dissected.
5. Forward FISH Analysis to Identify the Relative Position of the Components of the Dissected Chromosome
Steps
- Apply FISH analysis with the commercially available whole chromosome painting probes specific for each of the chromosome components, which are identified by microdissection to the GTG-banded metaphase spreads containing the dissected chromosome.
- Combining the G-banding and FISH results obtained from each probe determines the relative position of the components on the chromosome dissected.
Abeysinghe, H. R., Cedrone, E., Tyan, T., Xu, J., and Wang, N. (1999). Amplification of C-myc as the origin of the homogeneous staining region in ovarian carcinoma detected by micro-FISH. Cancer Genet. Cytogenet. 114, 136-143.
Bohlander, S. K., Espinosa, R., III, LeBeau, M. M., Rowley, J. D., and Diaz, M. O. (1992). A method for the rapid sequence-independent amplification of microdissected chromosomal material. Genomics 13, 1322-1324.
Carter, N. P., Ferguson-Smith, M. A., Perryman, M. T., Telenius, H., Pelmear, A. H., Leversha, M. A., Glancy, M. T., Wood, S. L., Cook, K., Dyson, H. M., Ferguson-Smith, M. E., and Willat, L. R. (1992). Reverse chromosome painting: A method for the rapid analysis of aberrant chromosomes in clinical cytogenetics. J. Med. Genet. 29, 299-307.
Guan, X.-Y., Trent, J. M., and Meltzer, P. S. (1993). Generation of band specific painting probes from a single microdissection chromosome. Hum. Mol. Genet. 2, 1117-11121.
Ludecke, H. J., Senger, G., Claussen, U., and Horsthemke, B. (1989). Cloning of defined regions of the human genome by microdissection of banded chromosomes and enzymatic amplification. Nature (Lond) 338, 348-350.
Ruano, G., Pagliaro, E. M., Schwartz, T. P., Lamy, K., Messina, D., Gaensslen, R. E., and Lee, H. C. (1992). Heat soaked PCR: An efficient method for DNA amplification with applications to forensic analysis. BioTech 13, 266-274.
Sen, S., Sen, P., Mulac-Jericevic, B., Zhou, H., Pirrotta, V., and Stass, S. (1994). Microdissected double-minute DNA detects variable patterns of chromosomal localization and multiple abundantly expressed transcripts in normal and leukemic cells. Genomics 19, 542-551.
Telenius, H., Carter, N. P., Bebb, C. E., Nordenskjold, M., Ponder, B. A. J., and Tunnacliffe, A. (1992). Degenerate oligonucleotideprimed PCR: General amplification of target DNA by a single degenerate primer. Genomics 13, 718-725.
Wang, H. C., and Federoff, S. (1972). Banding in human chromosomes treated with trypsin. Nature New Biol. 235, 52-54.
Wang, N. (2002). Methodologies in cancer cytogenetics and molecular cytogenetics. Am. J. Med. Genet. 115, 118-124.
Xu, J., Cedrone, E., Roberts, M., Wu, G., Gershagen, S., and Wang, N. (1995). The characterization of chromosomal rearrangements by a combined micro-FISH approach in a patient with myelodysplastic syndrome. Cancer Genet. Cytogenet. 83, 105-110.
Xu, J., Fong, C. T., Cedrone, E., Sullivan, J., and Wang, N. (1998). Prenatal identification of de novo marker chromosomes using micro-FISH approach. Clin. Genet. 53, 490-496.
Xu, J., and Wang, N. (1994). Identification of chromosomal structural alterations in human ovarian carcinoma cells using combined GTG-banding and repetitive fluorescence in situ hybridization (FISH). Cancer Genet. Cytogenet. 74, 1-7.
Zhang, M., Meltzer, P., Jenkins, R., Guan, X.-Y., and Trent, J. M. (1993). Application of chromosome microdissection probes for elucidation of BCR-ABL fusion and variant Philadelphia chromosome translocations in chronic myelogenous leukemia. Blood 81, 3365-3371.
Support our developers




