Confocal Microscopy of Drosophila Embryos
The genetically tractable organism Drosophila melanogaster is proving to be an excellent model system for cell biological analysis in the context of the whole organism. The relative ease with which embryos can be obtained in large numbers and processed for highresolution light microscopy has facilitated many recent advances at the interface between cell and developmental biology. Fine subcellular structures previously impossible to visualise by conventional fluorescence microscopy, on account of high noise resulting from out-of-focus signals, are revealed with clarity on a confocal microscope.
There are several reasons why scientists who have not used Drosophila before may wish to use Drosophila embryos for the analysis of protein localisation and expression. The embryo contains representatives of each cell type and is small enough (500 x 100 µm) to fit within the field of view of a 20X objective lens. The embryos are nearly transparent, permitting visualisation of all cells in whole mount preparations. These features allow one to assay the tissue distribution of a particular protein in a single specimen. The tissues have a relatively simple structure, with the epithelia being made up of a single layer of cells. In general, there are fewer copies of each protein encoded by the genome compared with vertebrates, e.g., one α-actinin rather than four, further simplifying the analysis of the distribution of a particular kind of protein. Injection of double-stranded RNA can be an effective way to knock down protein expression, provided the bulk of the protein in the embryo comes from new synthesis. Sophisticated manipulation of the proteins is possible using the powerful molecular genetic techniques available in this organism.
This article describes the techniques used to look at cell junctions and cytoskeletal structures in the developing embryo, using both antibodies against Drosophila proteins and GFP-tagged proteins expressed from transgenes.
A. Equipment
Fly bottles or vials (250-ml bottles and 30/40ml vials, Scientific Lab Supplies)
Egg collection cages (see Fig. 1, I)
Apple juice agar plates (see solutions)
Embryo collection baskets (see Fig. 1, I)
Scintillation vials (4ml, 908-054, Jencons; 20ml, 215/0079/00, BDH)
Rollers (Denley Spiramix 5 or equivalent)
Rotary shakers (Labinco BV or equivalent)
Microscope glass slides 76 x 26mm (Menzel Glaeser, Superfrost Color)
Coverslips 22 x 22mm, 22 x 40mm (Menzel Glaeser)
Forceps (Watkins and Doncaster)
A001 size dissecting tungsten needles (E6871, Watkins and Doncaster)
Needle holder (Watkins and Doncaster)
26G3/8, 0.45 x 10 syringe needle (Industrial and Scientific)
Artist's paintbrushes 00, 000
Bleach (commercial)
Heptane (27051-2, Sigma)
Methanol (M/4000/PC17, Fischer Chemicals)
Ethanol (E/0555DF/17, Fischer Chemicals)
Formaldehyde (37-40%, 101134-A, AnalaR/BDH, reagent grade)
Paraformaldehyde (40% EM grade, 15715, Electronmicroscopy Sciences)
Triton X-100 (T-9284, Sigma)
Bovine serum albumin (BSA) fraction IV (A-2153, Sigma)
NaCl (102415K, AnalaR/BDH)
KCl (101984L, AnalaR/BDH)
NaH2PO4 (301324Q, AnalaR/BDH)
Na2HPO4 (S-9390, Sigma)
KH2PO4 (102034B, AnalaR/BDH)
EGTA (E-4378, Sigma)
PIPES (P-8203, Sigma)
SDS (Ultrapure, Melford Labs Ltd.)
Glycerol (101184K, AnalaR/BDH)
Sucrose (S-0389, Sigma)
Apple juice (commercial)
Bactoagar (0140-01, Difco laboratories)
Nipagin (Nipa Laboratories Inc.)
Baker's yeast blocks (Commercial)
Vectashield (H-1000,Vector Labs, Burlingame)
Voltalef H10S Halocarbon oil (Elf Atochem, France)
Fly food (Instant Drosophila medium, Phillip Harris Scientific)
- 10× phosphate-buffered saline (PBS) (1 litre) (adapted from Sambrook and Russell, 2001): 80g (137mM final) NaCl, 2g (2.7mM final) KCl, 14.4g (10mM final) Na2HPO4, 2.4g (2mM final) KH2PO4, and distilled water up to 1 litre. Dissolve all components in 800ml of H2O and make volume up to 1 litre. Adjust pH to 7.4 with HCl/NaOH. Sterilise by autoclaving.
- PBT (500ml) PBS with 0.3% Triton X-100: 50ml 10x PBS, 750 µl Triton X-100 (0.3% v/v), 0.1 g (optional) sodium azide, and distilled water to 500ml
- PBTB (500ml) PBS with 0.3% Triton X-100 and 0.5% BSA: 50ml 10x PBS, 2.5g BSA (0.5%w/v), 750µl Triton X-100 (0.3% v/v), 0.1 g (optional) sodium azide, and distilled water to 500ml.
- PB: 77.4ml (100mM final) Na2HPO4 (1M) and 22.6ml (100mM final) NaH2PO4 (1M). Dissolve in distilled water to 1000ml. Adjust pH to 7.4.
- PEM (4 ml) (adapted from Ashburner, 1989): 400µl (100mM final) 1M PIPES (pH 6.9), 4µl (1mM final) 1M MgCl2, and 20µl (1mM final) 200mM EGTA (pH 8.0). Adjust to pH 6.9 with 10M KOH to final volume of 4ml.
- 4% formaldehyde in PBS (100ml): 10ml 10x PBS, 10.8 ml 37% formaldehyde, and 79.2ml distilled water.
- Apple juice agar plates (1.6 litre): 36g Bactoagar and 1200ml distilled water. Dissolve the agar in a microwave oven or on a heating pad until the solution is clear. Dissolve 40 g sucrose in 400 ml apple juice in a microwave oven or on a heating pad until the solution is clear. Mix the two solutions once clear on a stirrer. Add 20ml of 20% Nipagin when the solution has cooled below 60°C. Stir again and pour while still liquid into 5-cm/9-cm petri dishes. When set, store at 4°C. Allow to reach room temperature before use.
A. Producing Embryos for Confocal Microscopy
Steps
- Grow up flies. If you are not a fly laboratory, try to obtain a few bottles from a fly colleague. Alternatively, make simple bottles with instant fly food, topped with cotton or a foam plug. Try to get the density of flies such that they are neither under- nor overcrowded. For 250-ml bottles, 20 females and 10 males will lay the ideal number of eggs per bottle in a 24-h period, after an initial lag of a couple of days while they feed and mate. Such a bottle will produce about 500 flies after 10 days at 25°C, depending on the food and the fly strain. Ideally, collect adult flies within 1-2 days after eclosion from the pupal case. For the egg collection cages described later, 25-200 females and about half the number of males is a good number.
- Set up an egg collection cage containing an apple juice agar plate at the bottom, fixed on with tape, and a ventilation screen at the top (Fig. 1, IA). The plate should have an approximately 50 µl dab of yeast paste in the centre. Make yeast paste by stirring in a bit of water into the baker's yeast to make it the consistency of toothpaste. This is the food for the flies and is essential for the females to lay large numbers of eggs. Ensure that there is no excess moisture in the cage or plate (usually resulting from condensation on the plate). Put the flies into the cage and incubate at the desired temperature (usually 25°C). It will take a couple of days before the females lay substantial numbers of eggs, but once they get going each female can lay 100 eggs a day. Replace the yeasted apple juice plates at least once a day. To do this, invert the cage and peel back the tape holding the plate. Gently tap the flies down on the bench and quickly swap the plate. Even for the most practiced of us, this usually results in a few flies gaining freedom.
- Collect embryos for analysis. Place a new yeasted apple juice plate on the cage. After the appropriate time period (e.g., 4 hs), remove the plate from the cage, replacing with a fresh one, and age the plate containing the embryos at 25°C for the desired length of time. Such short collections are an advantage when one is interested in a particular developmental event (for staging, see Wieschaus and Nuesslein-Volhard, 1998; Campos- Ortega and Hartenstein, 1997), and for the novice who has not yet learned how to stage embryos by visual examination. Embryogenesis lasts 22h at 25°C, but the exoskeleton becomes secreted at about 16h, making embryos older than this inaccessible to antibody staining using standard methods. Development takes twice as long at 18°C, allowing some adjustment to more convenient hours. The embryo plates can also be stored at 4°C for up to 24h, but this has been known to cause subtle phenotypes. Overnight collections allow the examination of all stages of embryogenesis, but the distribution of embryos at the different stages is rarely even.
In this step, the embryos are collected from the plate, washed well to remove yeast and other detritus from the plate and the eggshell (chorion) removed. You should prepare vials ready for the fixation step (Cl) before starting this step.
Steps
- Add about 2ml water to each plate and, with the aid of a paintbrush, gently release the embryos from the agar (the eggs are usually partially pushed into the agar when they are laid).
- Rinse/pour embryos into a meshed basket, made as shown in Fig. 1, IB.
- Place the basket in a dish (the lids from the apple juice plates are useful for this) containing 50% bleach (1:1, water:commercial bleach). It should be made fresh and can be used for a maximum of 2 days. If older, the bleach still removes the chorion, but affects devitellinisation adversely. Swirl the basket in the dish and add more bleach into the basket. After 2-5 mins, most embryos will have risen to the surface, indicative of successful dechorionation. Do not extend the period in bleach beyond 5min; prolonged exposure can destroy tissue architecture.
- Wash the embryos thoroughly while in the basket by swirling embryos under running tap water or water from a squirt bottle.
- Briefly dry embryos by placing them on a paper towel. Do not overdry.
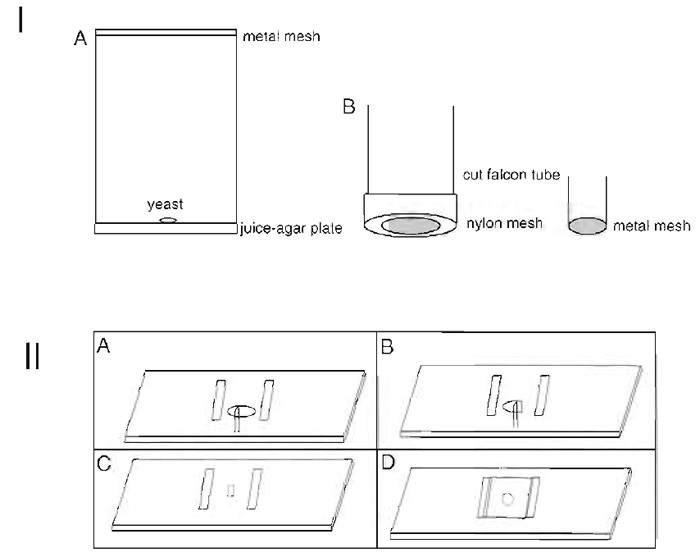 |
| FIGURE 1 Schematic showing the parts that make up an egg-laying cage (IA). Apple juice agar is poured and allowed to set in petri plates. A dab of yeast is placed on warmed plates that are placed on top of cages containing flies. The plate is strapped onto the cage and the contraption is placed inverted in an incubator. The parts that make up an egg collection basket are shown in IB. A nylon mesh/sieve is screwed onto a cut 50-ml Falcon tube by the cap in which a hole has been cut using a hot scalpel blade. A 15-ml cut Falcon tube can also be used to make smaller baskets using metal wire mesh that is cut into rounds of the appropriate diameter and fastened onto the tube by melting the end of the tube with a hot scalpel blade. (II) Schematic showing how sections are cut. See text for details. |
Steps
- Label a scintillation vial for each different sample of embryos (stage or genotype) and add 0.8ml of 4% formaldehyde in PBS.
- Transfer the basket of embryos into a dish containing heptane. Using a Pasteur pipette, transfer the embryos with the heptane into the scintillation vial containing 4% formaldehyde in PBS. The amount of heptane should be equal to or greater than the volume of fixative solution. Mix for 20-30mins at room temperature or at 37°C on a roller or other device.
- Remove the aqueous phase from the bottom with a Pasteur pipette and then remove the heptane. Add 1ml of new heptane and then rapidly add 1-3 ml of methanol and shake by vigorous inverting/vortexing for 1 min. (If you want to stain the embryos with phalloidin, use 80-90% ethanol rather than methanol at this and the next two steps; this has the disadvantage that reduced number of embryos are successfully devitellinised.)
- Let the embryos settle for about 10s and then transfer the embryos from the bottom of the vial to an Eppendorf tube with a Pasteur pipette. Any embryos still at the interface between the heptane and the methanol have not popped out of the vitelline membrane and therefore will not stain well and should be left behind. An alternative method for this step is remove all of the liquid phase, including embryos that have not settled, leaving only the devitellinised embryos at the bottom of the vial. Add 1-2ml of methanol to the embryos and vortex/shake for 30s. Remove methanol.
- Wash in methanol three times. For each wash, squirt in 1ml of methanol so that the embryos disperse. Let the embryos settle by gravity and remove the methanol (this is called a gravity wash). Either proceed to the next step or add 1ml methanol and store at -20°C (for embryos that will be stained with phalloidin store in 100% ethanol).
Embryos can be stored in methanol or ethanol at -20°C for several months and will continue to be successfully stained by most antibodies. However, detection of some antigens and GFP fluorescence are affected adversely by exposure to ethanol or methanol. In these cases, do steps 3-5 as rapidly as possible and proceed quickly to step D1. Some antibodies will not recognise their antigen in embryos fixed by this method, but will when one of the alternative methods described in Section III, G is used.
D. Antibody Staining of Embryos
Steps
- Wash embryos in PBT (PBS with 0.3% Triton X-100): First give three to five quick washes, where 1ml is squirted in so the embryos are dispersed, the embryos are allowed to settle by gravity, and then the PBT is removed (a gravity wash) and then give one longer wash (15mins) with mixing. We use a device that gently inverts the tubes for this and all subsequent steps, but a variety of mixing devices should work. If mixing is not sufficient, it will result in variable staining of embryos in the tube.
- (Optional) Staining with some antibodies is improved by a 15-min incubation in 0.1% SDS with mixing, followed by three washes in PBT. This may help those antibodies raised against bands from SDS gels.
- Block nonspecific binding sites on the embryos by incubating in PBTB for 30min. Transfer 15µl (5- 20µl) of embryos to a 0.5-ml Eppendorf tube for each staining with a Pipetteman and a yellow tip with the last couple of millimeter of the end cut off with a razor blade. Rinse the tip with some PBT before taking up the embryos. While the volume of embryos is not critical, larger volumes can result in uneven staining. The volume of 15µl of embryos corresponds to about 100 embryos, which is sufficient for most purposes. If more embryos are desired, use multiple tubes or a larger tube for staining.
- Incubate embryos with 250-500µl of PBTB containing an appropriate dilution of each primary antibody at 4°C overnight. Optimal timing depends on the antibody and whether staining of internal tissues is important, in which case longer incubations are recommended. We have successfully used a range from 2h at room temperature to 2 days at 4°C. If the appropriate dilution is not known, a good starting point is 1 :1000 for antisera and 1:10 for monoclonal antibody supernatants.
- Wash off the primary antibody with PBT with three to five gravity washes and one to three longer (at least 15 min) washes.
- Incubate with fluorochrome-conjugated secondary antibodies diluted in PBTB as suggested by the manufacturer (usually 1:100-300) at room temperature for 2h or overnight at 4°C. The choice of fluorochrome coupled will depend on the laser lines of the confocal you are using. Our preferred ones are Alexa488, Alexa568 or Cy3, and Cy5. We tend to keep the embryos in the dark during the incubation.
- Wash secondary antibodies off with three to five gravity washes and three longer (5-30min each) washes in PBT. Washing overnight is also fine. The embryos can be left for a day or two in PBT at 4°C, but it is better to store them after step F1 or once mounted on microscope slides.
E. Staining with Phalloidin to Visualise Actin
Steps
- Embryos that have been exposed to methanol will not stain with phalloidin, so use 90% ethanol rather than methanol during devitellinisation.
- Dissolve rhodamine-conjugated phalloidin in methanol (methanol harms the actin, not the phalloidin) and store at -20°C in aliquots of 6 units. To stain embryos, vacuum dessicate the required number of aliquots and resuspend in PBTB at 1 unit/100µl.
- Follow sections D1 and D2. (If phalloidin staining is to be combined with antibody staining, proceed through sections D3 to D5 and then go to E4. Phalloidin can be combined with the secondary antibodies at D6, at the concentration described later.
- Incubate embryos with 1 unit of phalloidin (100 µl of the aforementioned solution, plus 400 µl PBT or PBTB)/0.5 ml tube containing approximately 20µl of embryos for at least 30min.
- Wash twice with PBT (if combined with secondary antibodies, wash more thoroughly).
- After the final wash, replace PBT with 50-200µl of Vectashield or similar glycerol-based mounting medium containing antibleaching agents. The embryos can be stored at 4°C for a couple of days or, for longer periods, allow 30min to equilibrate in the Vectashield and store at -20°C.
- Using a Pipetteman and a yellow tip with the end cut off, transfer some of the embryos onto a slide. First suck up approximately 5µl of Vectashield alone and then the embryos, as this reduces sticking of the embryos to the inside of the yellow tip. For a 22 x 22-mm coverslip, use 20-30µl per slide. The lower volume will lead to a mild flattening of the embryos, which can be an advantage, but is trickier to mount without air bubbles. As you add the embryos to the slide, move the tip to spread them out over a square region a bit smaller than the coverslip. Using a needle or similar instrument (such as plastic Pipetteman tip with a very small diameter), gently distribute the embryos more evenly and separate them from each other. (Do not fiddle too long with this, as they will generally distribute into a monolayer when the coverslip is placed on top, but this step helps reduce or eliminate the number of embryos lying on top of each other.) The goals of the experiment will dictate the optimal number of embryos per slide. For staged embryos, it is better to have fewer embryos, less than 30 per slide. You can then in a single session on the confocal completely examine all embryos on the slide and start a new session with a new slide. This is helpful because it is easier to find the best embryos at low power and then add oil for the image collection. If you have embryos from an overnight collection, a larger number of embryos improves the chance of finding each stage on a given slide. With a forceps, gently place a coverslip over the embryos, starting at one end to avoid introducing air bubbles and gently let go of the forceps as the Vectashield spreads. If a small volume has been used to flatten the embryos, let sit for 10min and then if there is still air under the coverslip, add a little more Vectashield to an edge.
- (Optional) Seal the edges with nail varnish (but see Section III,G,5).
General Comments
Almost every aspect of this procedure will vary from fly laboratory to fly laboratory and even within our laboratory there are a number of variations. None of the following are critical: which recipe is used for the egg collection plates (some laboratories prefer grape juice), the tubes used for fixation and staining, the solutions used for antibody staining, the timing of the incubations and the number of washes, and the medium for mounting the embryos. As mentioned at the beginning, the key steps are fixation and devitellinisation. Again there are many variants of this, and the important thing is to get a method that works well for the antibodies you are using. Once you have a method that works, try to do this step as identically as possible each time. This will give you reproducibly good staining. Generally, the first time a new person in the laboratory does this technique it does not work well. However, after a few times, it is hard to understand how you could have failed the first time. A simple check is to look at a few embryos at step C4 or D1 down a dissecting microscope. If the embryos are still surrounded by the transparent shiny vitelline membrane, throw them away and try again. Before moving onto microscopy, we will mention some useful variants of this technique.
1. Alternative Fixation Solutions In these the method is identical to that described earlier, but instead of 1:1 4% formaldehyde in PBS: heptane, and 20-30min at room temperature, use the following alternatives.
1:1 4% formaldehyde in PEM:heptane, 20-30min at room temperature.
1:1 4% formaldehyde in PB :heptane, 20-30 minutes (Uemura et al., 1996).
1:1 37% formaldehyde:heptane, 2-5min at 25-37°C with gentle mixing
This can improve the preservation of cytoskeletal structures present in membrane compartments [e.g., scribble, β-heavy spectrin (Bilder et al., 2000)] and gives the best preservation of microtubules (Foe et al., 2000).
2. Heat Fixation (Muller et al., 1996)
Heat fixation improves the penetration of antibodies into late stage embryos and larvae (when the exoskeleton normally blocks antibodies).
Step
- With a paintbrush, transfer dechorionated embryos from the dechorionation basket into a scintillation vial containing 1-2ml of 0.4% NaCl and 0.3% Triton X-100 solution that has been heated in a boiled, but no longer boiling, water bath with its cap half screwed on.
- Pull the vial out of the hot water bath, screw cap tightly, and shake once vigorously.
- Uncap the vial and fill up with ice-cold 0.4% NaCl and 0.3% Triton X-100.
- Leave on ice until cooled.
- Pour off the solution, add heptane and methanol in equal volumes, and vortex to devitellinize.
- Remove embryos from bottom, transfer to new tube, and wash twice with methanol.
- Incubate in methanol for an additional hour and proceed as usual for antibody staining.
Steps
- Transfer dechorionated embryos to 4ml of fixation solution: 1 volume 40% formaldehyde (EM grade) and 3 volumes PBS.
- Vortex for 45 s and add 4ml heptane.
- Shake vigorously for 25 min.
- To devitellinise the embryos, use either 80-90% ethanol (as for phalloidin described earlier) or hand peel embryos after replacing the fixation solution with PBS many times (see Section III,J,1).
4. Methanol Fixation
Steps
- Transfer dechorionated embryos to 1:1 heptane: (97% methanol, 3% 0.5 M NaEGTA, pH 8.0)
- Shake hard for 2min.
- Transfer embryos from the bottom of the vial to a 1.5-ml Eppendorf tube.
- Wash three times with methanol
- Incubate in methanol overnight at 4°C.
This is a good alternative when a new antibody fails to stain embryos fixed under standard conditions, as a number of antibodies have been found to only work on embryos prepared by this method. Other antibodies will work with both this method and standard fixation, while some will not stain with this method.
If you are planning to visualise GFP fluorescence, keep exposure of the embryos to methanol or ethanol to a minimum and do not seal the slides with clear nail varnish. Organic solvents quench GFP fluorescence or severely affect its ability to fluoresce.
H. Embryo Thick Sections (Adapted from Grosshans and Wieschaus, 2000)
To get a good cross-sectional view of the embryo by confocal microscopy it is necessary to cut sections. They give far better resolution than optical X-Z scans (see later and Figs. 2E and 2F). We have used a relatively crude method that does not require a microtome and is compatible with our standard methods for antibody staining. These sections are particularly useful for the visualisation of morphogenetic events that occur in the midline and in the interior of the embryo, such as invagination of the mesoderm, formation of the midgut, and dorsal closure.
- Fix and stain the embryos according to the desired protocol. It is hard to combine this method on phalloidin-stained embryos, because the ethanol (rather than methanol) treatment results in embryos that are harder to cut and the morphology is not as well preserved.
- Follow the antibody staining protocol described in Section III, C. Keep embryos in PBT after the secondary antibody has been washed off.
- Refix the stained embryos in 4% formaldehyde in PBT for 30-60min at 37°C.
- Wash the embryos in PBT, do three gravity washes, and one 15-min wash with mixing.
- Take the embryos through a glycerol series comprising of 10, 20, and 30% glycerol in PBT for 1h each followed by 40% overnight. (This is the optimal concentration, do not go higher.)
- On clean dusted microscope slides, make bridges with strips of coverslips cut using a diamond knife. These are stuck to the slide using clear nail varnish.
- Place an embryo on the slide in a drop of Vectashield between the bridges (see Fig. 1, IIA).
- To cut the embryo, use a 26G3/8 hypodermic needle fitted to a 2.5-ml syringe whose plunger has been removed. Cut at one end in one clean sweep with the bevelled edge facing away from the slice you wish to keep. Turn the embryo around with the dissecting needle. Once again, with the bevelled edge of the needle pointing away from the slice you want to keep, cut the embryo to obtain a slice of the desired region (Fig. 1, IIB). The thickness of the section must be approximately the same thickness as the bridges; that is the thickness of a coverslip, which is about one-fifth the length of the embryo. If the sections are thinner, they are hard to orient and most of the section is beyond the depth of focus of the 60x objective; if they are thicker, the morphology is distorted by the resulting squashing. With practice you can cut more than one section from the same embryo, but each should be mounted on a separate slide due to slight differences in thickness.
- Remove the undesired embryo pieces with the dissecting needle (Fig. 1, IIC) and orient the desired slice so that it lies at the bottom of the drop of Vectashield with its anterior face up.
- Place a 22 x 22-mm coverslip over the section and the bridges (Fig. 1, IID). Do not seal the sides to allow for reorientation. If the section is slightly thinner than the thickness of the coverslip, it may move/turn in the drop of Vectashield. Slight pressure on the corner of the coverslip can then bring the section back to its right orientation. If the section moves under the coverslip during imaging, abandon that section and proceed to the next.
- View under a 60x oil immersion objective lens.
Drosophila embryos can be examined with a variety of confocal microscope setups. Both inverted and upright microscopes are appropriate. The key variable is the available laser lines; this will dictate the optimal choice of fluorochrome-conjugated secondary antibodies that one should use. A detailed description of confocal microscopy is clearly beyond the scope of this article (a good reference is Centonze and Pawley, 1995), but we hope to provide a few helpful hints.
Once one has obtained an initial quick view of the result of the staining, which is often possible by conventional fluorescence microscopy if the signal is strong, the usual goal is to produce a set of images for publication. The biggest challenge is to produce a set of comparable images of different embryos. For example, for a general description of the distribution of a new protein one would like comparable views of embryos at different stages; generally a lateral view. There is a strong convention in the fly field regarding images of embryos: they should always have anterior to the left and dorsal up. Similarly, for a comparison of staining in different mutants, it is essential to obtain a suitable micrograph from wild type and each mutant at the same stage and with the same view: this can often require looking through many embryos. Clearly, to be able to achieve this you must be able to tell the stage of the embryo and which way is up, which is not as easy as it sounds. The only way to achieve this is to spend time looking at embryos down the microscope. To learn your way around embryonic development, it helps to examine embryos that are stained with antibodies that highlight the development of a particular tissue. The following antibodies are recommended for this: Fasciclin III (available from the Developmental Studies Hybridoma Bank) and phosphotyrosine (available from Sigma). For an overall view of the embryo, phalloidin (rhodamine conjugated, Molecular Probes) is very useful (see Fig. 2).
In general, we start our confocal session by first scanning through the slide by conventional fluorescence, taking note of the approximate position on the slide of embryos of the right stage and orientation that have stained well. Next we find a well-stained embryo that is not one of the best ones and use it to work out the appropriate settings for each channel on the confocal. When it is essential that there is minimal mixing between the outputs of each channel, then it is advisable to do sequential scanning rather than simultaneous. Use the channel with the brightest signal to get started. In most cases we use a collection area of 1024 x 768 pixels. Using rapid scanning, find the best focal plane, adjusting the focus by hand (adjustments to the eyepieces often result in the view by eye being at a different focal plane than the resultant scan). Using intermediate scan rates, set the laser power, iris, gain, and background levels appropriately for the first channel and then proceed to the next channel. We keep the iris similar in the different channels so that they all capture the same depth of focus. Some confocal setups have this feature built into the software.
Once all channels are set up, adjust the focus to obtain the optimal image. Collect images using Kalman scanning to improve the signal-to-noise ratio. We use either 166 lines per second with three to five scans or 50 lines per second and two scans. Make a note of the orientation of the embryo, especially if you are just focused on a small region.
One other variable to keep an eye on is the zoom. We standardly keep this at 1 (no zoom), and it is helpful to check that it has not been varied by a previous worker, as your images from session to session will not be comparable. Some judicious use of the zoom function can improve resolution. In practice we have found that a zoom factor of greater than two provides no further improvement in resolution and may even lead to loss of resolution.
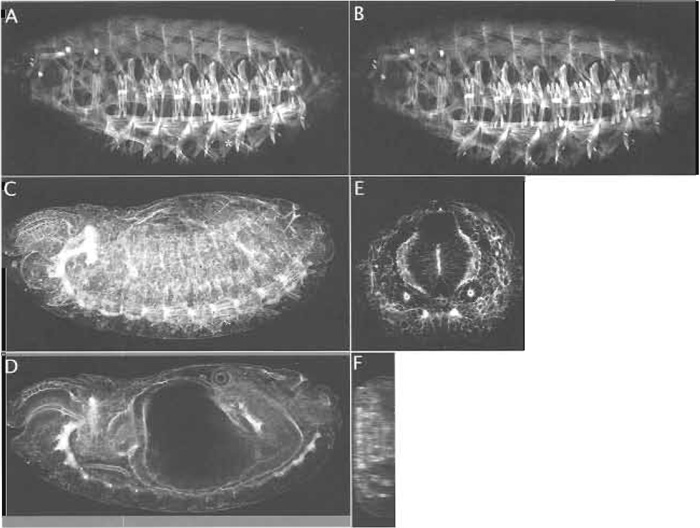 |
| FIGURE 2 A projection of four optical sections of a late Drosophila embryo stained with phalloidin to highlight actin in somatic muscles (A) is compared to a single optical section (B). Note that some muscles (asterisk in A) are not visualised in the single optical section. A projection of 42 sections of a Drosophila embryo stained with phalloidin is used to visualise actin in the whole embryo (C). A single optical section through the same embryo (D) reveals staining in internal structures that are obscured by the extensive surface staining in C. A confocal generated X-Z section along the Z axis depicted by the white line in D is shown in F. Such a scan provides very poor resolution. A single optical section of a thick section cut (as described in Section III,H) to include the same region of the embryo as shown in D is shown in E. Such a section resolves the apicobasal extension of actin especially in internal organs that are obscured in whole embryos. In all images, the dorsal surface of the embryo is on top. In A-D, the anterior end of the embryo is on the left. |
Steps
- For imaging live embryos, briefly dry embryos over a paper towel after dechorionation. Alternatively, the embryos can be dechorionated by hand by gently rolling on double-stick 3MM Scotch tape with a forceps.
- Transfer embryos in their vitelline membranes with a paintbrush or forceps to a drop of halocarbon oil. Cover with a coverslip and leave the slides unsealed. For time-lapse analysis it is essential to use a method that allows better access of air to the embryo. We use a microscope slide consisting of an airpermeable Teflon membrane in a holder (Edwards et al., 1997; Kiehart et al., 1994).
IV. COMMON PROBLEMS
A. Nonstaining Embryos
The most common causes are (i) use of inappropriate fixation method, (ii) inefficient devitellinisation, (iii) inappropriate primary and/or secondary antibody, and (iv) faulty laser, either nonfunctioning or unsuitable for the fluorochrome coupled to the secondary antibody.
B. Bleaching
This is a common problem inherent to confocal microscopy, especially during the acquisition of a Z series at high laser power and high magnification. It can be reduced by ensuring that the laser power is set to the minimum value that is optimal for the image.
C. Low-Resolution Images
Low-Resolution Images can result from faulty image processing of the original image. Obtain the highest resolution image possible on the confocal. Information that does not exist in the acquired data cannot be added by image processing software. Too high a gain can result in too much noise. Such noisy images may be improved by increasing either the iris or the laser power. Out of focus signals resulting from inappropriate mounting and from inappropriate thickness of sections are the cause of low-resolution images from sections.
Acknowledgments
We thank Sylvia Erhardt, Anja Hagting, Jasmin Kirchner, Jordan Raft, Katja Roeper, Alex Sossick, and Christos Zervas for comments and suggestions.
Ashburner, M. (1989). "Drosophila, a Laboratory Manual," p. 372. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Bilder, D., Li, M., and Perrimon, N. (2000). Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289(5476), 113-116.
Campos-Ortega, J., and Hartenstein, V. (1997). "The Embryonic Development of Drosophila melanogaster," pp. 1-102. Springer- Verlag, Berlin.
Centonze, V., and Pawley, J. (1995). Tutorial on confocal microscopy and use of the confocal test specimen. In "Handbook of Biological Confocal Microscopy" (J. Pawley. ed), pp. 549-569. Plenum Press, New York.
Davis, I. (1999). Visualising fluorescence in Drosophila: Optimal detection in thick specimens. In "Protein Localization by Fluorescence Microscopy" (J. V. Small, ed.), pp. 133-161. Oxford Univ. Press, Oxford.
Edwards, K. A., Demsky, M., Montague, R. A., Weymouth, N., and Kiehart, D. P. (1997). GFP-moesin illuminates actin cytoskeleton dynamics in living tissues and demonstrates cell shape changes during morphogenesis in Drosophila. Dev Biol. 191, 103-117.
Foe, V. E., Field, C. M., and Odell, G. M. (2000). Microtubules and mitotic cycle phase modulate spatiotemporal distributions of Factin and myosin II in Drosophila syncytial blastoderm embryos. Development 127(9), 1767-1787.
Grosshans, J., and Wieschaus, E. (2000). A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell 101(5), 523-531.
Kiehart, D. P., Montague, R. A., Rickoll, W. L., Foard, D., and Thomas, G. H. (1994). High resolution microscopic methods for the analysis of cellular movements in Drosophila embryos. Methods Cell Biol. 44, 507-532.
Muller, H. A., and Wieschaus, E. (1996). armadillo, bazooka, and stardust are critical for early stages in formation of the zonula adherens and maintenance of the polarized blastoderm epithelium in Drosophila. J Cell Biol. 134(1), 149-163.
Rothwell, W. F., and Sullivan, W. (2000). Fluorescent analysis of embryos. In "Drosophila Protocols" (M. Ashburner, W. Sullivan, and R. Scott Hawley, eds.), pp. 141-158. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Sambrook, J., and Russell, D. W. (2001). "Molecular Cloning: A Laboratory Manual," A1.7. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Uemura, T., Oda, H., Kraut, R., Hayashi, S., Kotaoka, Y., and Takeichi, M. (1996). Zygotic Drosophila E-cadherin expression is required for processes of dynamic epithelial cell rearrangement in the Drosophila embryo. Genes Dev. 10(6), 659-671.
Wieschaus, E., and Nuesslein-Volhard, C. (1998). Looking at embryos. In "Drosophila, a Practical Approach" (D. Roberts, ed.), pp. 179-213. IRL Press (Oxford Univ. Press), Oxford.
Support our developers




