Cytoskeleton Proteins
The visualisation of cytoskeletal proteins and their dynamics in live cells has led to invaluable insights into cytoskeleton-driven processes as various as cell migration, chromosome segregation during division, cytokinesis, and phagocytosis, as well as the establishment of cell-substrate or cell-cell adhesion. The cytoskeleton of eukaryotic cells can be grossly divided into three filamentous systems: actin filaments, microtubules, and intermediate filaments.
This article focuses on revealing the dynamics of the actin cytoskeleton and the microtubule system, as well as some proteins associating with them in interphase cells. It compares classical approaches with more recent advancements in this enormously growing field.
More specifically, this article discusses visualisation of the cytoskeleton using purified components chemically modified with fluorescent dyes and compares this method with ectopic expression of cytoskeletal genes fused to fluorescent protein tags such as green fluorescent protein (GFP). While the efficient introduction of fluorescently labelled cytoskeletal proteins is usually achieved by microinjection, which requires additional equipment and experimental effort, the expression of genes tagged to fluorescent proteins is obtained by transfection of the respective fusion constructs. We will show that both methods can lead to equally satisfying results, but as the transfection of nucleic acids seems to be continuously improved and developed further, the latter method may be used more frequently for most common tissue culture cells. However, because studies on cytoskeleton dynamics are sometimes performed on cell types that have so far not been reported to mediate the expression of ectopic genes, such as epidermal fish keratocytes, protein microinjection and subsequent analysis are still performed. In addition, we briefly mention the microinjection of specific antibodies to monitor the dynamics of cytoskeletal components.
II. MATERIALS AND INSTRUMENTATION
A. Equipment
1. Microscope
Inverted microscope (e.g., Axiovert 135TV, Carl Zeiss Jena GmbH) equipped for epifluorescence and phase-contrast microscopy with 40x/1.3NA and 100x/1.4NA oil immersion objectives, 1.6 and 2.5 optovar intermediate magnification, electronic shutters (e.g., Uniblitz Electronic 35-mm shutter including driver Model VMMD-1, BFI Optilas) to allow for computer-controlled opening of the light paths, filter wheel (e.g., LUDL Electronic Products LTD, SN: 102691 and driver SN: 1029595) to enable two-colour epifluorescence in combination with appropriate dichroic beam splitters and emission filters (Omega Optical Inc. or Chroma Technology Corp.), and tungsten lamps (Osram, HLX64625, FCR 12V, 100W) for both phase contrast and fluorescence light paths. Tungsten lamps are not as bright as mercury lamps, but the latter cause photodamage and bleaching with higher probability.
2. Data Acquisition
Preferably by a back-illuminated, cooled chargecoupled- device camera (e.g., Princeton Research Instruments TKB 1000 x 800, SN:J019820; Controller SN:J0198609) driven, for instance, by IPLab (Scanalytics Inc.) or Metamorph software (Universal Imaging Corporation). Dependent on cell types and/ or processes studied, this "epifluorescence setting" can be extended to the various options of confocal microscopy (see contributions on confocal imaging within this volume).
Commercial microinjection capillaries (Femtotips I; Cat. No.: 5242952.008, Eppendorf AG) or, alternatively, self-made needles requiring a "needle puller" (e.g., Model PN-30, Narishige International LTD), mechanical (Cat. No.: 520137, Leica Microsystems), oilpressure- driven (e.g., Model M0188NE, SN:99069, Narishige International LTD), or electronic (e.g., Model 5171, Eppendorf AG) micromanipulators used in combination with an air pressure device (e.g., Transjector 5246, Eppendorf AG); flexible microloaders for loading microinjection capillaries (Cat. No.: 5242956.003, Eppendorf AG).
4. Centrifuges
Cooled high-speed benchtop centrifuges (e.g., Biofuge fresco, Heraeus) to pellet protein aggregates prior to injection.
5. Heating
Open (e.g., Series 20 chamber platform, Model PH4, Warner Instruments used with heater controller model TC-324B, SN:1176) or, alternatively, closed heating chambers (e.g., Model FCS2, Bioptechs Inc.).
Cells growing at room temperature are observed on coverslips mounted to the bottom of plastic dishes harbouring a central hole or in comparable commercial devices.
6. Coverslips
Round glass coverslips, 15 or 40mm in diameter, cleaned in 6/4 ethanol/HCl (37%), washed extensively with H2O, dried, and sterilised by exposure to ultraviolet light or in a dry heat sterilizer at 220°C (ethanol: Sigma, Cat. No.: E-7023; HCl: Sigma, Cat. No.: H-7020).
General equipment and plastic-ware (e.g., from Becton Dickinson Biosciences or Greiner Bio-One GmbH) for molecular biology techniques and tissue culture.
B. Cells and Media
1. Cells
Mouse melanoma cells B16-F1 (American Type Culture Collection: CRL-6323) and goldfish fin fibroblasts (CAR, American Type Culture Collection: CCL-71).
2. Growth
All reagents are from Invitrogen Corp. unless stated otherwise. For B16-F1: Dulbecco's modified Eagle's medium (Cat. No.: 41965-039) supplemented with 10% fetal calf serum (FCS, PAA Laboratories, Clone, Cat. No.: All-041), 2mM glutamine (Cat. No.: 25030-024), 1% antibiotics (Cat. No.: 15070-063). For CAR: basal Eagle's medium with Hank's balanced salt solution (Cat. No.: 21370-028), 15% serum (Hyclone, Cat. No.: SH30070.03), 1mM glutamine, 1mM nonessential amino acids (Cat. No.: 11140-035), and 1% antibiotics. Upon confluence, cells are detached using trypsin (Cat. No.: 25300-054) and seeded according to standard protocols.
3. B16 Medium for Microscope
Ham's F12 HEPES-buffered medium (Sigma, Cat. No.: N8641) including complete supplements of the regular growth medium (see point 2).
4. Substrates
Laminin (Sigma, Cat. No.: L2020) or fibronectin (Roche, Cat. No.: 1051407).
5. Lipofection
With Superfect transfection reagent (Qiagen GmbH, Cat. No.: 301305) or FuGENE6 (Roche, Cat. No.: 1 814 443).
- TAMRA-vinculin: From turkey gizzard, coupled to carboxytetramethylrhodamine (5'-TAMRA) succinimidyl ester (Molecular Probes, Cat. No.: C-2211) as described (Rottner et al., 1999) and upon addition of 2mg sucrose/mg protein stored in 30-µl aliquots at 1 mg/ml at -70°C
- TAMRA-α-actinin: From turkey gizzard, kindly provided by M. Gimona (Salzburg) and fluorescently modified and stored as described for vinculin
- Cy3-tubulin: As described (Hyman, 1991); for a protocol of tubulin labelling (by John Peloquin) we recommend http://www.borisylab.nwu.edu/pages/protocols/cy3tub.html
- EGFP-tubulin: Kindly provided by M. Geese (Braunschweig), comprises murine β3-tubulin fused into EGFP-C2 (Clontech, Cat. No.: 6083-1)
- EGFP-EBI: As described (Stepanova et al., 2003)
- EGFP-p16-B: p16B-cDNA, also known as ARPC5B (Millard et al., 2003), was amplified from a human EST clone (Acc. No.: 12652556, RZPD cloneID IRALp962P167) and subcloned into EGFP-C1 (Clontech; Cat. No.: 6084-1)
- Zyxin-EGFP: As described (Rottner et al., 2001)
- Zyxin-dsRED: As described (Bhatt et al., 2002)
III. PROCEDURES
Solutions
All chemicals are from Sigma-Aldrich.
- Phosphate-buffered saline (PBS) working solution: 140 mM NaCl (Cat. No.: S-7653), 2.7 mM KCl (Cat. No.: P-1338), 10mM Na2HPO4 (Cat. No.: S-7907), 1.8mM KH2PO4 (Cat. No.: P-0662), pH 7.4
- Urea: 2M in H2O (Cat. No.: U-0631)
- Laminin coating buffer: 50mM Tris (Cat. No.: T- 1503) adjusted with HCl to pH 7.5, 150mM NaCl
- Microinjection buffer (for vinculin and α-actinin): 2 mM Tris-acetate (acetic acid: Cat. No.: A-0808), 50 mM KCl (Cat. No.: P-1338), 0.1mM dithioerythritol (DTE, Cat. No.: D-8255), pH 7.0. Note that microinjection buffers vary significantly depending on the cytoskeletal protein injected [e.g., buffers for microinjecting actin generally lack KCl (Wang, 1984), whereas buffers for injecting myosin II require KCl (up to 450mM) in order to avoid polymerisation of the respective component in the needle (Verkhovsky and Borisy, 1993)].
- Microinjection buffer (for tubulin): 80mM PIPES (Cat. No.: P-6757) adjusted with KOH (Cat. No.: P- 6310) to pH 6.8, 1 mM MgCl2 (Cat. No.: M-2670), 1 mM EGTA (Cat. No.: E-1644) (Fig. 1).
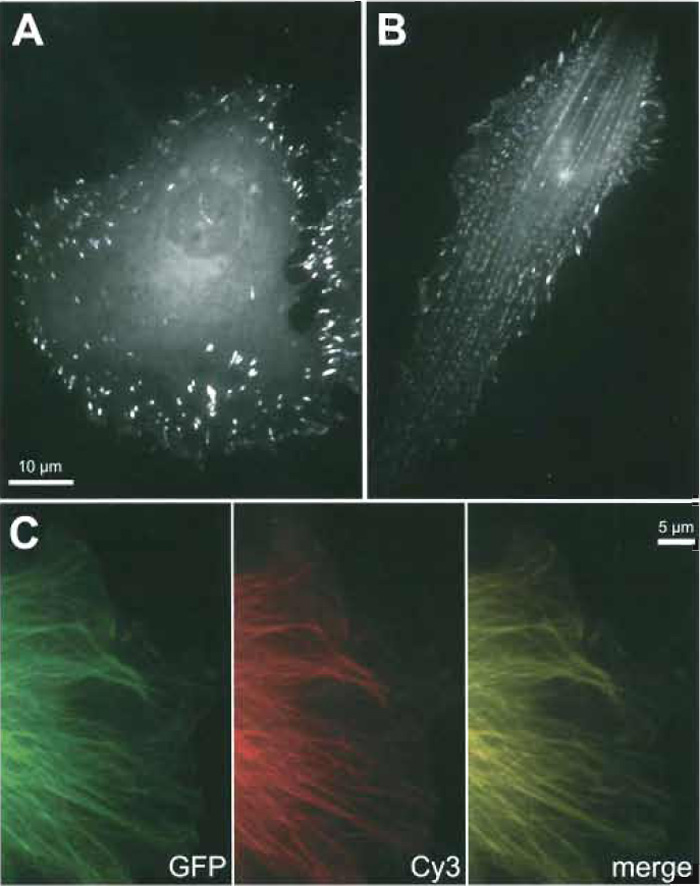 |
| FIGURE 1 Examples for specific incorporation of fluorescently tagged, microinjected proteins: CAR fish fibroblasts microinjected with TAMRA-vinculin (A) or TAMRA-α-actinin (B). Note the specific recruitment of both probes to focal adhesions and the additional periodic incorporation of α-actinin into stress fibres. (C) EGFP-β-tubulin (kind gift of M. Geese, Braunschweig) expression (green) in a CAR cell additionally microinjected with Cy3-tubulin (kind gift of F. Severin, Dresden; red). Note that both probes reveal an identical microtubule pattern (merge) and can therefore be used equally to study microtubule dynamics. |
A. Visualisation Using a Fluorescently Conjugated Protein
Steps
- Various different fibroblast cell lines survive microinjections better when grown on fibronectin. For coating of coverslips, dissolve fibronectin at a concentration of 1 mg/ml in 2M urea and store at 4°C.
- Dilute fibronectin stock 1:20 to 50µg/ml with PBS and coat sterile coverslips for 1 h at room temperature (150µl for 15-mm-diameter coverslips). Before seeding the cells, wash thoroughly with PBS to remove urea (two to three times).
- Upon washing, seed the cells onto coverslips in a petri dish and let them attach and spread for 12- 72h in an incubator.
- Thaw an aliquot of fluorescently coupled protein. If not in appropriate buffer, dialyse the sample into microinjection buffer using Slide-A-Lyzer (e.g., Pierce, 10 kDa cutoff Cat. No.: 66415) or a similar device for the dialysis of small volumes and concentrate using microcon concentrators (Amicon, YM-10, Cat. No.: 42407) (for proteins such as vinculin or α-actinin, concentrations of 0.5-1mg/ml are ideal for injections).
- Spin the probe for microinjection in a cooled centrifuge at maximum speed (approximately 10,000g) for at least 30min before loading the needle in order to remove protein aggregates that may clog up the needle tip.
- Mount cells onto the microscope in a chamber freely accessible to the microinjection needle.
- Upon loading of the needle from the back using a flexible pipette tip, carefully inspect the needle tip for the absence of air bubbles. If required, remove them by gently hitting the needle shaft, but proceed rapidly in order to avoid drying of the tip.
- Apply pressure (20-100 hPa) to the needle holder, attach needle, and rapidly move needle into the medium. (The "background pressure" applied before entering the medium is required to avoid sucking up of the medium due to capillary force of the needle tip.)
- Bring the needle close to the cells.
- Before injections, check the needle flow by fluorescence.
- In case of flow, gently inject the cells, manually or using a so-called half-automatic system (Eppendorf AG, see earlier discussion). For manual injections, carefully approach the membrane and remove immediately upon needle flow into the cell.
- Upon injection of a sufficient number of cells, allow the cells to recover and the cytoskeletal proteins to incorporate (e.g., 30-60min). For actin, full incorporation into the cytoskeleton may take up to 2h. In contrast, vinculin targets to focal adhesions within minutes.
- After incorporation of the injected protein, darken the room and search for interesting cells using epifluorescence and preferably a 40x oil immersion lens. In case no fluorescent cells can be found by eye, it will be difficult to pick up a satisfying signal using a back-illuminated CCD camera.
- After selection of an interesting cell, switch to higher magnification (100x) with or without optovar intermediate magnification and test for the minimal exposure time required to get a good signal/noise ratio. (For important details on fluorescence imaging and image processing, see articles by Anderson and by Wang.)
- In order to record a stack of fluorescent images, set the appropriate number of images to be taken and the time period in between individual frames (dependent on experiment). In order to avoid photodamage, illuminate cells with strong fluorescent light as rarely as possible. Avoid focusing in between frames using epifluorescence; focus using transmitted light, for example, in phase contrast, because for transmitted optics, much lower lamp intensities are required.
Steps
- Purify the expression vector for your GFP-tagged open reading frame of interest using standard DNA purification columns (e.g., Qiagen-tip 500); dilute the construct in H2O in concentrations of not less than 0.5 µg/µl.
- Seed the cells into 3.5-cm-diameter dishes (Falcon, Cat. No.: 35.3001) and allow them to attach and spread for 12-24h.
- Transfect cells in 3.5-cm dishes 12-24 h according to standard protocols, e.g., using FuGENE (Roche; 3 µl FuGENE/µg DNA) or Superfect transfection reagent (Qiagen; 6µl Superfect/µg DNA). In our hands, 1-2 µg of DNA is sufficient per 3.5-cm-diameter dish.
- Detach transfected cells using trypsin and seed onto coverslips. For studying the dynamics of GFPtagged proteins during the actin-based motility of B16- F1 melanoma cells, we coat coverslips with 25µg/ml laminin in laminin-coating buffer (1 h at room temperature); laminin stock (1mg/ml) is stored in small aliquots at -20°C.
- Upon spreading (for B16-F1 cells, wait 3-5h), mount coverslips on the microscope using an appropriate open or closed chamber system, including a heating device if required. Compensate for lack of CO2 by using HEPES-buffered growth media if needed. Changing to HEPES-buffered media may require some adaption time (dependent on cell type).
- Darken the room and search for transfected cells with your eyes with epifluorescence preferably using a 40x/1.3NA oil immersion lens.
- Upon identification of an interesting cell, proceed as in steps 13 and 14 in Section III,A.
IV. GENERAL COMMENTS ON FLUORESCENT TAGGING
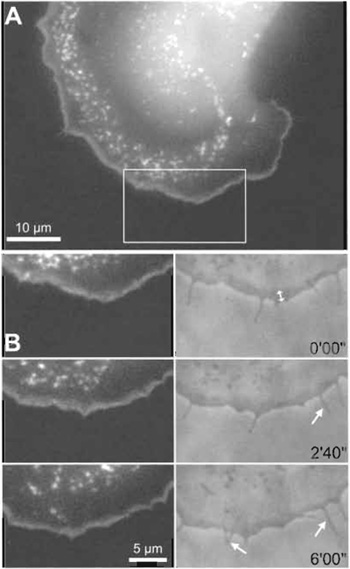 |
| FIGURE 2 Arp2/3 dynamics in motile B16-F1 melanoma cell as revealed by transient expression of the GFP-tagged novel isoform p16B (ARPC5B). (A) Note that EGFP-p16B incorporated into the lamellipodial actin meshwork at the cell periphery and into highly dynamic surface ruffles, also known as actin flowers or clouds, as expected. (B) The time-lapse sequence from the region boxed in A (right panel: phase contrast) reveals reorganisation of the Arp2/3 complex during advancement of the cell periphery. Time is in minutes and seconds. Note the relatively constant lamellipodium width (double-headed arrow in B) during forward movement and the virtual exclusion of this Arp subunit from protruding filopodia (arrows). |
For the fusion of cDNAs to fluorescent protein tags, the most common of which currently is the enhanced green fluorescent protein (EGFP, Clontech), also known as GFPmutl (Cormack et al., 1996), we recommend always comparing the fusion of the given cDNA to both the N and the C terminus of the tag because it is difficult to predict whether the fusion to the protein tag on either side may interfere with protein function. For instance, in order to generate reliable fusion proteins for the visualisation of the actin nucleating complex Arp2/3 (Higgs and Pollard, 2001), we have so far generated N- and C-terminal fusions to each of the seven components and tested their incorporation in cells, with varying results. Some of the constructs showed poor incorporation or even interfered with lamellipodia protrusion (p34-EGFP). Good results were obtained with small molecular weight components of the complex (p16 and p21), although both Nand C-terminally tagged Arp3 also proved useful (Stradal et al., 2001). Figure 2 shows the dynamics of p16B (ARPC5B), a novel isoform of p16 (Millard et al., 2003) fused to the C terminus of EGFP in B16-F1 cells. In addition, spacers between EGFP and the gene of interest can be crucial for incorporation of the fusion constructs (Geese et al., 2000).
V. CHOICE OF COMBINATIONS FOR DUAL LABELING
As with conventional fluorescent dyes, fluorescent protein tags today also come in different colours. As opposed to multiple labellings of fixed samples (see article by Prast et al.), for live cell imaging as described here, only dual-labelling experiments have proven practical so far, probably due to the lack of sufficiently bright dyes excitable by UV excitation. We provide examples here for imaging EGFP-tagged proteins together with injected proteins tagged to rhodamine derivatives or to Cy3 (see Fig. 3). An important alternative is the use of the red fluorescent protein from Discosoma sp. (dsRED), exemplified here by a dsREDzyxin probe combined with EGFP-tubulin (Fig. 4). However, in contrast to EGFP, dsRED and even the improved variant termed dsRED2 (Clontech) are known to form a tetramer when fluorescent (Baird et al., 2000). Unfortunately, this feature can interfere with the function of certain cytoskeletal proteins. For instance, expression of a dsRED-calmodulin fusion protein was reported to cause severe aggregation and therefore mislocalisation of the tagged protein (Mizuno et al., 2001).
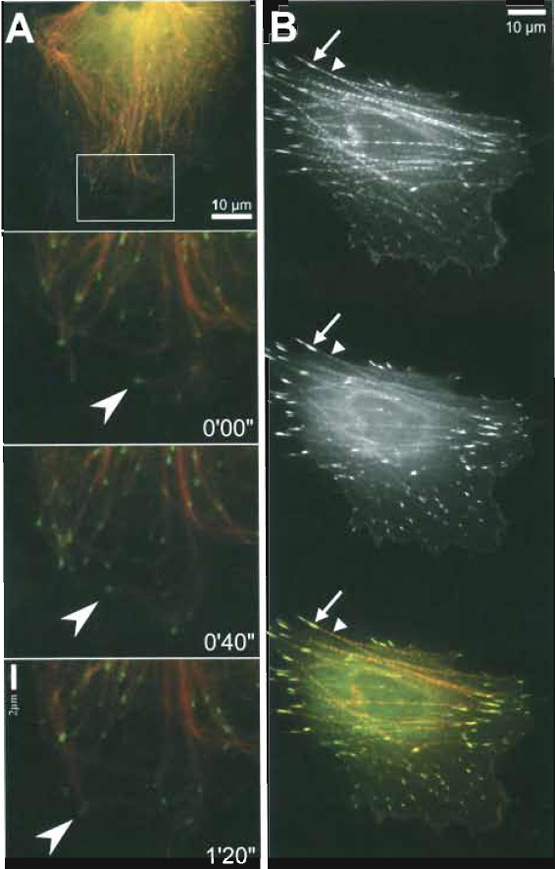 |
| FIGURE 3 Examples of simultaneous visualisation of two distinct cytoskeletal components in the same cell. (A) Dynamics of microtubule plus ends in fish fibroblast CAR. Microinjected Cy3-tubulin (red) combined with ectopically expressed EGFP-EB1 (kind gift of A. S. Akhmanova, Rotterdam; green). Top image, overview. Bottom images, zoom into the boxed region in the overview, subsequent frames, time shown in minutes and seconds. Note that EB1 localises to the tips of growing microtubules (e.g., arrowheads in frames 0'00" and 0'40"), but is absent from pausing or shrinking microtubule ends (e.g., arrowhead in frame 1'20"). (B) EGFP-zyxin expressing CAR cell microinjected with TAMRA-α-actinin: As shown in Fig. 2B, the α-actinin probe (top panel and red in the merged image at the bottom) mainly incorporates along stress fibers (arrowheads) and in focal adhesions (arrows). Zyxin (middle panel and green at the bottom), thought to be recruited to the cytoskeleton via α-actinin binding (Reinhard et at., 1999), shows a similar, although not identical distribution. The merged image (bottom) reveals that zyxin (green) is enriched in focal adhesions, whereas α-actinin (red) targets more prominently to stress fibres, indicating that the subcellular positioning of zyxin is more complex than simple α-actinin-mediated recruitment. |
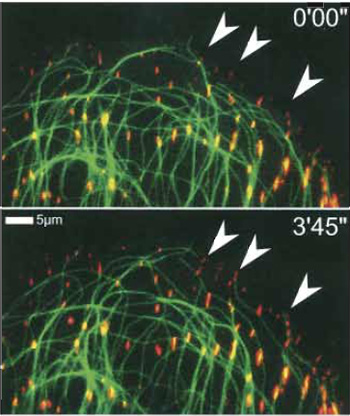 |
| FIGURE 4 Simultaneous visualisation of two cytoskeletal components using two distinct fluorescent protein tags. Combination of EGFP-β-tubulin (pseudocoloured red) and dsRED-zyxin (kind gift of A. Huttenlocher, Madison; green) expressed ectopically in a CAR fish fibroblast. The combination reveals the formation and microtubule targeting of focal adhesions on this slowly protruding cell edge. Subsequent frames, time shown in minutes and seconds. Note newly formed adhesions (frame 3'45") and microtubules associated with them (arrowheads). |
Exciting more recent developments include far red fluorescent proteins, which appear separable not only from EGFP, but also from ECFP and EYFP. One was obtained from mutating a nonfluorescent chromoprotein from Heteractis crispa, termed HcRED-2A in (Gurskaya et al., 2001), and is available as HcRED-1 (Clontech). The protein is a dimer and can now also be obtained as a "monomeric" tandem variant (evrogen.com).
In addition, Tsien and colleagues have developed a true monomeric red fluorescent protein (termed mRFP1) with excitation and emission peaks at 584 and 607, respectively, by introducing multiple mutations into dsRED (Campbell et al., 2002). Comparisons of HcRED-1, its tandem variant, and mRFP1 as fusion proteins with actin and other cytoskeletal components revealed that mRFP1-at least in fusion with the components tested-was superior to the former red variants, mainly due to the lack of aggregation (not shown). Hence, although about 25% of the brightness of the original dsRED, the mRFP1 probe in combination with EGFP is expected to prove very useful for live cell dual imaging.
We thank Drs. Mario Gimona and Fedor Severin for kindly providing purified smooth muscle proteins and Cy3-tagged tubulin, respectively, Dr. R. Y. Tsien for mRFP1 cDNA, Drs. Anna S. Akhmanova, Marcus Geese, and Anna Huttenlocher for expression constructs, and Petra Hagendorff for excellent technical assistance.
References
Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2000). Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 97, 11984-11989.
Bhatt, A., Kaverina, I., Otey, C., and Huttenlocher, A. (2002). Regulation of focal complex composition and disassembly by the calcium-dependent protease calpain. J. Cell Sci. 115, 3415- 3425.
Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2002). A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 99, 7877-7882.
Cormack, B. P., Valdivia, R. H., and Falkow, S. (1996). FACSoptimized mutants of the green fluorescent protein (GFP). Gene 173, 33-38.
Geese, M., Schluter, K., Rothkegel, M., Jockusch, B. M., Wehland, J., and Sechi, A. S. (2000). Accumulation of profilin II at the surface of Listeria is concomitant with the onset of motility and correlates with bacterial speed. J. Cell Sci. 113(Pt. 8), 1415-1426.
Gurskaya, N. G., Fradkov, A. F., Terskikh, A., Matz, M. V., Labas, Y. A., Martynov, V. I., Yanushevich, Y. G., Lukyanov, K. A., and Lukyanov, S. A. (2001). GFP-like chromoproteins as a source of far-red fluorescent proteins. FEBS Lett. 507, 16-20.
Heim, R., and Tsien, R. Y. (1996). Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 6, 178-182.
Higgs, H. N., and Pollard, T. D. (2001). Regulation of actin filament network formation through ARP2/3 complex: Activation by a diverse array of proteins. Annu. Rev. Biochem. 70, 649-676.
Hyman, A. A. (1991). Preparation of marked microtubules for the assay of the polarity of microtubule-based motors by fluorescence. J. Cell Sci. Suppl. 14, 125-127.
Mizuno, H., Sawano, A., Eli, P., Hama, H., and Miyawaki, A. (2001). Red fluorescent protein from Discosoma as a fusion tag and a partner for fluorescence resonance energy transfer. Biochemistry 40, 2502-2510.
Reinhard, M., Zumbrunn, J., Jaquemar, D., Kuhn, M., Walter, U., and Trueb, B. (1999). An alpha-actinin binding site of zyxin is essential for subcellular zyxin localization and alpha-actinin recruitment. J. Biol. Chem. 274, 13410-13418.
Rottner, K., Hall, A., and Small, J. V. (1999). Interplay between Rac and Rho in the control of substrate contact dynamics. Curr. Biol. 9, 640-648.
Rottner, K., Krause, M., Gimona, M., Small, J. V., and Wehland, J. (2001). Zyxin is not colocalized with vasodilator-stimulated phosphoprotein (VASP) at lamellipodial tips and exhibits different dynamics to vinculin, paxillin, and VASP in focal adhesions. Mol. Biol. Cell 12, 3103-3113.
Stepanova, T., Slemmer, J., Hoogenraad, C. C., Lansbergen, G., Dortland, B., De Zeeuw, C. I., Grosveld, F., van Cappellen, G., Akhmanova, A., and Galjart, N. (2003). Visualization of microtubule growth in cultured neurons via the use of EB3- GFP (end-binding protein 3-green fluorescent protein). J. Neuroscience 23, 2655-2664.
Verkhovsky, A. B., and Borisy, G. G. (1993). Non-sarcomeric mode of myosin II organization in the fibroblast lamellum. J. Ceil Biol. 123, 637-652.
Wang, Y. L. (1984). Reorganization of actin filament bundles in living fibroblasts. J. Cell Biol. 99, 1478-1485.
Support our developers




