Direct Immunogold Labeling of Components within Protein Complexes
Immunolabeling has long been applied to the study of localization and distribution of antigens within cells and tissues. Perhaps the most exciting recent development, however, is in the use of gold-conjugated antibody labeling in localizing the position of subcomponents in macromolecular complexes. As metal clusters are small and dense and can be specifically targeted to residues within molecular complexes, they can significantly enhance the structure-function information obtained from electron micrographs of macromolecular complexes. Colloidal gold can be prepared in sizes ranging from lnm to over 30nm (e.g., Baschong et al., 1985; Slot and Geuze, 1985), allowing a simultaneous detection of several antigens within the same electron microscope preparation.
Gold particles can either be linked covalently to fortuitously placed reactive groups within, or added to, the protein complex under study or be conjugated with primary or secondary antibodies, which in turn bind to exposed epitopes on proteins, so-called indirect immunolabeling. When whole cells or ultrathin sections are labeled using an indirect immunogold labeling method, the distance between the colloidal gold and the epitope is normally below the level of resolution of these techniques. However, in terms of labeled protein complexes in a transmission electron microscope, the large distance between gold and antigen-binding site can be problematic in specifically localizing the epitope in question. The size of an antibody is about 15nm, therefore the distance of a gold particle (centre of the particle) from the epitope will still be of the order of about 30 nm, even when a small gold particle such as lnm of gold is used. To identify a subunit within a complex of about 40 to 50nm, such as cytoplasmic dynein, it is necessary to bring the gold particle much closer to the epitope. Use of a goldconjugated primary antibody will bring the gold particle within a range of about 15 nm. Further improvement in resolution can be achieved when the colloidal gold is bound to an Fab fragment, obtained by a papain digest of IgGs (Mage, 1980).
This chapter article details our methods of preparing colloidal gold-conjugated primary antibodies (Fab fragments), which, in combination with a modified technique of negative staining, can be employed to identify subunits within protein complexes.
1,2-Dichloroethane (VWR), bacitracin (Sigma, B0125), borax (Sigma, B3545), bovine serum albumin (BSA, Sigma, A7030), carbon rods (Agar, E416), Centricon- 30 microconcentrators (Amicon), centrifuge rotor (Beckman, 80Ti), centrifuge tubes (Nalgene, 3118-0010), chloroform (VWR), column (MoBiTec, 2.5ml), dimethylchlorosilane (Sigma, D6258), dithiothreitol (DTT, Sigma, D5545), EDTA (Sigma, E9884), filter paper (Whatman, 1001-150, 1004-150), forceps (Agar, T5268), Formvar (Agar, R1201), glass microscope slides (Agar, L4242), glycerol (Sigma, G6279), gold chloride (Sigma, G4022), grid storage boxes (Agar, G276A), grids (Taab Lab's), iodoacetamide (Sigma, I6125), lens tissue (EMS), magnetic stirrer (Agar, G3784), methanol (VWR), mica sheets (Agar, G250-1), Oak Ridge centrifuge tubes (Nalgene, 3118-0010), papain (Sigma, P4742), petri dishes (VWR, 402/0062/06, 02/0066/01), potassium carbonate (Sigma, P5833), potassium chloride (Sigma, P9333), protein G (Sigma, P3296), protein G-Sephadex (Sigma, S 1235), razor blades (Agar, T5016), Schott bottles (VWR, 215/0150/10), sodium azide (Sigma, S8032), sodium chloride (Sigma, S7653), sodium phosphate (Sigma, S8282, S7907), sodium thiocyanate (Sigma, S7757), microprobe sonicator (Jencons-PLS, Sonics Vibracell), staining jar (Agar, L4108), staining jars (Sigma, S6016), staining trough (VWR, 406/0230/10), ultracentrifuge (Beckman, LE80K), uranyl acetate (Agar, R1260A), and carbon evaporation unit (Edwards 306).
A. Preparation of Specimen Support for Transmission Electron Microscope
Ideally, very thin but stable support films are required for high-resolution transmission electron microscopy; however, this is problematic as very thin films are extremely fragile. It is for this reason that more robust holey films with an overlay of ultrathin carbon only over the holes are often used. The methodology is as follows.
1. Preparation of Holey Formvar Film
Solutions and Equipment (Materials)
- 0.5% (w/v) Formvar in chloroform (freshly prepared)
- 50-ml Schott bottle (wide neck)
- 50% (w/v) glycerol in dH2O
- Forceps, self-closing
- Glass staining trough spray-painted black of 7cm depth and 12 cm width (or larger)
- Grids, 300-mesh copper Maxataform HF35
- Lens paper, lint free
- Magnetic stirrer
- Methanol
- Microprobe sonicator
- Microscope slides
- Petri dishes, glass, 150mm diameter
- Razor blades
- Staining jar (Coplin)
- Whatman filter papers #1; #4, qualitative circles, 150mm diameter
- Freshly prepare 50ml Formvar solution in a Schott bottle; stir to dissolve with magnetic stirrer for about 15-20 min.
- Prepare fresh 50% glycerol solution and add 15-20 drops to dissolved Formvar; shake vigorously for 30 s. Sonicate for 2 min at full power using a microprobe sonicator. Probe should be placed one-third from bottom of solution in a 50-ml Schott bottle. The amount of glycerol and sonication time will vary depending on size and density of holes required. The bottle should feel warm after sonication.
- Place Formvar-glycerol solution into a staining jar. Clean several microscope slides with lint-free lens tissue. Dip slides one at a time into the Formvar solution, remove rapidly and vertically to ensure an even film, stand on their ends on filter paper, and let dry completely. (Note: Prepare all of the slides at one time because the dispersion of glycerol solution is unstable and the droplets slowly coalesce.)
- Test the film quality. As soon as a slide is dry, float off the film (see later), pick up a couple of grids (see step 6 for details), dry briefly, and check in EM for hole size, number, and distribution. The holes should be just larger than the photographic field at the desired working magnification.
- Fill glass staining trough to the brim with clean dH2O. Wipe surface with sheet of lint-free lens tissue.
- Score edges of a slide with clean single-edged razor blade. Breath lightly onto the slide and then, with that side uppermost, carefully float off the film on to the water surface by slowly sliding the slide into the water at a 45° angle. Place grids on film, rough (dull) surface down using self-closing forceps. Cut a rectangular piece of #4 Whatman filter paper approximately 30% larger than the floated plastic film; slightly bend the ends up to use as handles. Pick grids and film up with filter paper rectangle by gently touching the paper horizontally to floating Formvar film. Place the strip of filter paper on more filter paper to dry. After the grids are dry, place filter paper/grids/Formvar on a bed of #1 filter paper saturated with methanol in a glass petri dish for 10min. This "etching" procedure removes any glycerol/water droplets and dissolves any very thin plastic lying across the holes.
- After the grids are dry, check a grid in the scope to ensure the spread and size of holes.
- Place filter paper with grids in a glass petri dish and store in a refrigerator.
Do not overload floating Formvar film with grids, as it will sink! As a rough guide, use no more than 25-30 grids per slide-sized piece of Formvar, neither should one place grids too close to the edge of the plastic. Reflected light from a lamp and/or a dark surface underneath the glass dish will help in observing films clearly. Because plastic films tend to be unstable in the electron beam, the holey Formvar can be substituted by holey carbon film to obtain more stable grids as follows. This is particularly important for high-resolution work.
2. Coating of Holey Formvar Grids with Thick Carbon for Additional Stability
Solutions and Equipment
- 1,2-Dichloroethane
- Carbon evaporation unit (Edwards 306)
- Carbon rods, spectrographically pure
- Grid storage box
- Petri dishes, glass, 150mm diameter
- Place filter paper with grids in a carbon evaporator.
- Coat grids heavily with carbon at a vacuum in the range 10-6 Torr.
- Place filter paper/grids/Formvar/carbon on a bed of filter paper saturated with 1,2-dichloroethane in a glass petri dish for 2h. This removes the Formvar, leaving the holey carbon. Remove and dry in a glass petri dish.
- Store grids in a grid storage box. They are stable for several months.
3. Preparation of Ultrathin Carbon Support Films on Mica Sheets
Solutions and Equipment
- Carbon evaporator unit
- Carbon rods, spectrographically pure
- Mica sheets, 75 × 25 mm
- Petri dishes, plastic, 100mm diameter
Steps
- Freshly cleave mica sheets. Keep cleaved side clean; do not touch with fingers at any time!
- In a carbon evaporator, expose mica sheets with new surface towards the carbon source. Coat mica sheets at a vacuum of 1 × 10-6 Torr or better with thin (about 10nm) carbon film (Note: Gradual deposition over several evaporations produces stronger and better quality carbon film.) Avoid sparking during carbon deposition to ensure a smooth deposition.
- Remove coated mica sheets from carbon evaporator, place in a plastic petri dish, and store in a refrigerator (careful, very delicate).
From some mica brands, carbon may not float off easily. Cleave mica just before coating and do not expose to air for longer than absolutely necessary. Position a screw on the corner of the filter paper so that a shadow will fall on the paper when the carbon is evaporated. The area where the paper is shielded from the carbon source will appear light compared to the rest helping to judge thickness.
B. Digest of IgG with Papain (Fab)
The protocols to obtain Fab fragments vary depending on the type and isotype of antibody used. We have used the protocol described here successfully to generate Fab fragments from rabbit IgG and mouse IgG1.
Solutions and Equipment
- 1mM DTT
- 100mM EDTA
- 1M iodoacetamide (freshly prepared)
- 5 mg/ml papain (can be stored as aliquots at -80°C)
- Phosphate-buffered saline (PBS): 150mM NaCl, 2.7 mM KCl, 50mM phosphate, pH 7.5
- Protein G-Sephadex column
- Adjust antibody concentration to 1-2mg/ml with PBS and add up to 2 mM EDTA and up to 1 mM DTT (depending on IgG type, use β-mercaptoethanol, DTT, or glycine as reducing agent).
- Prepare a 5-mg/ml papain stock solution. Please note that some commercial papain does not dissolve readily and will therefore yield a much lower level of digestion.
- Add papain to antibody solution; IgG:papain = 100 mg: 1 mg.
- Incubate at 37°C for 30-60min.
- Stop reaction by adding up to 30mM iodoacetamide and incubate in the dark on ice for 60min.
- Dialyze against PBS at 4°C.
- Check efficiency of digest by SDS-PAGE (12% gel under reducing conditions).
- Test antibody by immunochemistry.
- Remove Fc from Fab via protein G or protein A column (use protocol according to manufacturer).
Comments and Pitfalls
In our view the most reliable method to generate Fab fragments is by using a papain digest; however, care should be taken when choosing the reduction medium and the source of papain as there can be a considerable variation in potency of the reaction. Other enzymes, which can be used to cleave antibodies, include ficin and pepsin (e.g., Mage, 1980; Marianai et al., 1991). Please also note that protein A may not be used with all IgG isotypes and that some mouse IgG might bind to protein G via the Fab portion rather than the Fc.
Colloidal gold can be prepared easily in different sizes (e.g., Baschong and Wrigley, 1990; Slot and Geuze, 1985). We describe here our adaptation of the method by Baschong et al. (1985) to prepare 2-3 nm colloidal gold.
Solutions for Gold Preparation
- 0.2M K2CO3, 0.22 µm filtered
- 1% (w/v) HAuCl4 in dH2O
- 1.0M NaSCN, 0.22 µm filtered
- 200-ml siliconized Erlenmeyer flask
Steps [Modified According to Baschong et al. (1985)]
- Siliconize an Erlenmeyer flask by rinsing it with a 5% solution of dimethylchlorosilane in chloroform in fume hood, rinse with water after evaporation of solvent in a fume hood, and bake at 180~ for 2h.
- Add 0.5ml of 1% HAuCl4 to 100ml dH2O in a siliconized Erlenmeyer flask followed by 0.75ml of 0.2M
K2CO3 to gold solution while stirring. - Finally add 0.3ml of 1M NaSCN while stirring (note that the solution will become yellowish).
- Leave solution in a dark place overnight and use the following day for binding to antibodies.
In order to bind a single gold particle per IgG or Fab fragment it may be necessary to experiment in varying the protein colloidal gold ratio and/or adding varying amounts of an unspecific protein, e.g., BSA (Baschong and Wrigley, 1990) or bacitracin (Steffen et al., 1997). Newly developed gold probes from Nanoprobes (Stony Brook, NY), e.g., the 1.4nm colloidal gold within an organic shell, allow the coupling of the gold probe covalently to an antibody.
Solutions for Gold Labeling of the Antibody
- 10mg/ml bacitracin
- 10mg/ml BSA
- 10% (w/v) NaCl
- 10% (w/v) NaN3 (kept in a dark bottle)
- 10% (w/v) sucrose in PBS
- 10X TBS: 100mM Tris-HCl, 1.5M NaCl, pH 7.4
- 10-ml Oak Ridge centrifuge tubes
- 2 mM borax
- 5% dimethylchlorosilane in chloroform
- Centricon-30 microconcentrator
- Fume hood
- PBS: 150mM NaCl, 2.7mM KCl,10mM phosphate, pH 7.5
- Spectrophotometer
- Ultracentrifuge, 80Ti rotor
- Dialyse antibody into 2 mM borax or use Centricon microconcentrator for buffer exchange.
- Adjust colloidal gold solution to pH 8.0.
- Determine the minimum amount of antibody needed to stabilize the gold.
- Prepare a serial dilution of antibody from 100 to 5 µg IgG per 100 µl dH2O.
- Add 0.5 ml colloidal gold solution and incubate for 2min.
- Add 0.5ml of 10% NaCl and incubate for 30min.
- Measure the turbidity (formation of aggregates) using a spectrophotometer at OD550nm. (An increase in turbidity will mark the minimal concentration to stabilize gold particles.)
- Add antibody at determined minimal concentration (usually about 20 µg/ml) to colloidal gold solution while stirring and incubate on ice for 15 min.
- Add 100µg/ml bacitracin or BSA to cover the free surface of gold particles and incubate on ice for 60 min.
- Add 1/10th of volume of 10X TBS and centrifuge through a 1-ml 10% sucrose cushion in a Beckman 80Ti rotor at 47,000rpm (150,000gave) for 30min.
- Aspirate supernatant and resuspend soft pellet in about 200gl TBS containing 0.02% NaN3 and store in a refrigerator.
The 2- to 3-nm colloidal gold requires some time to develop. However, if the colloidal gold is stored for an extended period of time, it tends to form aggregates. We normally stabilise to colloidal gold with a protein solution, i.e., antibody, 12 to 24h after initiation of the reduction of the gold solution.
E. Negative Staining of Immunogold-Labeled Protein Complexes
A hydrophilic carbon support film is critical in achieving uniform high-quality negative staining, but carbon films tend to become more hydrophobic over time when exposed to air. The carbon film can be exposed to ionised gas (glow discharge) to revitalize its hydrophilic property. In our method (see also Marchese et al., 1995), negative staining is carried out on a carbon surface that has never been exposed to air, thus maintaining the hydrophilic nature of the carbon. This is achieved by floating the carbon film first onto the protein solution and then onto uranyl acetate. An example of an immunogold-labelled protein complex is illustrated in Fig. 1.
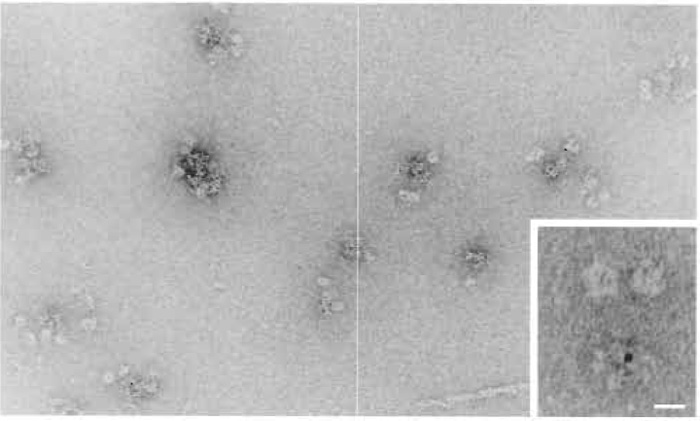 |
| FIGURE 1 Immunogold labeling of cytoplasmic dynein. The image shows the formation of clusters caused by the antibody-gold complex. In this case the gold was stabilised primarily by the presence of antibodies. Cytoplasmic dynein with a single gold particle could be observed (insert), if gold probes were used, where the antibody was supplemented with a nonspecific protein during the antibody-gold complex formation. Magnification bar in insert 10 µm. |
- 1% (w/v) uranyl acetate in dH2O (kept in the dark)
- 1.5-ml Eppendorf reaction vials, painted black
- Carbon-coated mica (see earlier discussion)
- Forceps (several pairs of self-closing, fine-tipped forceps)
- Grids with holey carbon film or holey Formvar film (see earlier discussion)
- Single-edged razor blades
- Small, adjustable angled lamp
- Whatman filter paper #1
Steps
- Paint the outside of a 1.5-ml Eppendorf reaction vial black.
- Mix gold-conjugated primary antibody with isolated purified protein complex and incubate at room temperature for 10 min.
- Dilute protein complex-antibody mixture to about 10 to 20µg/ml and place about 250µl of the mixture into the cap of a 1.5-ml Eppendorf reaction vial (Fig. 2, step 1).
- Fill the reaction vial with freshly filtered (0.22-µm syringe filter) 1% uranyl acetate. Note that the meniscus of the staining solution should be slightly above the rim of the reaction vial (Fig. 2, step 1).
- Reflect light from a lamp off the surface of the liquid at an angle of about ~45°
- Cut a thin carbon-coated mica sheet into about 3 x 5-mm pieces and score the carbon film with a clean single-edged razor blade about 2mm from one side of the small mica sheet.
- Using a pair of self-closing, fine-tipped forceps, partially float off the thin carbon film from the supporting mica sheet onto the surface of the protein solution (Fig. 2, step 2).
- Withdraw the carbon film after 2-3 s and float the carbon film completely free of the mica onto the surface of the staining solution (Fig. 2, step 3).
- Pick up the carbon film immediately with a 300- mesh grid coated with a holey Formvar or holey carbon film. [Note: Use a small adjustable angle light source to help visualise the thin carbon by reflection (Fig. 2, step 4)].
- Remove excess uranyl solution by touching edge of grid with the edge of a torn filter paper (Fig. 2, step 5).
- Let dry in a humid chamber (Fig. 2, step 6).
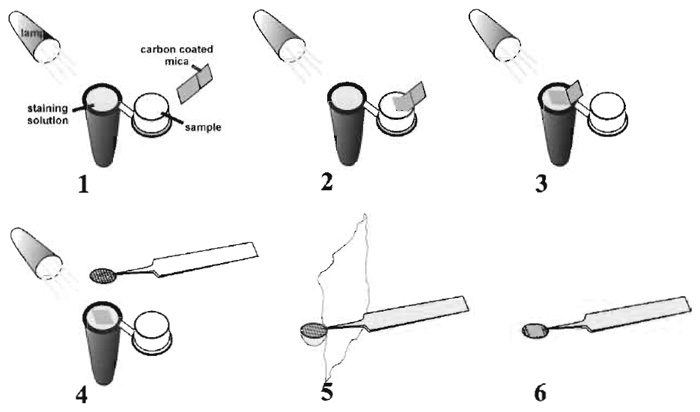 |
| FIGURE 2 Diagram illustrating the procedure for negative staining of protein complexes. See text for details of the procedure. |
Understanding the structural organization of a large protein complex is of increasing importance in our aim to understand their functional properties. Threedimensional image reconstruction by single particle analysis has proven to be a useful tool to achieve this goal. A potential development in the use of immunogold- conjugated antibodies is in the high-resolution three-dimensional image reconstruction of macromolecular complexes, allowing for the specific localisation of functional epitopes within molecular complexes. Technological developments in the production and specificity of heavy metal clusters (Nanoprobes Inc.), and advances in transmission electron microscopy and image analysis have enabled for the orientation of actin subunits within actin filaments to be investigated (Steinmetz et al., 1998). Thus far such studies have only been carried out with gold particles directly (covalently) linked to the molecule of interest via incorporated His tags (Hainfeld et al., 1999) or fortuitously exposed, or added, cysteines (Zlotnick et al., 1997). In the case of antibodies, disulphide bonds can be employed to attach a gold particle at a specific site, thereby making single epitope labelling more reliable.
References
Baschong, W., Lucocq, J. J., and Roth, J. (1985). "Thiocyanate gold" small (2-3 nm) colloidal gold for affinity cytochemical labelling in electron microscopy. Histochemistry 83, 409-411.
Baschong, W., and Wrigley, N. G. (1990). Small colloidal gold conjugated to Fab fragments or to immunoglobulin G as high-resolution labels for electron microscopy. J. Electron Microsc. Technol. 14, 313-323.
Hainfeld, J. F., Liu W., Halsey, M. R., Freimuth, P., and Powell, R. D. (1999). Ni-NTA-gold clusters target his-tagged proteins. J. Struct. Biol. 127, 185-198.
Mage, M. G. (1980). Preparation of Fab fragments from IgGs of different animal species. Methods Enzymol. 70, 142-150.
Marinai, M., Camagna, M., Tarditi, L., and Seccamani, E. (1991). A new enzymatic method to obtain high-yield (Fab)2 suitable for clinical use from mouse IgG1. Mol. Immunol. 28, 69-77.
Steffen, W., Hodgkinson, J. L., and Wiche, G. (1996). Immunogoldlocalisation of the intermediate chain within the protein complex of cytoplasmic dynein. J. Struct. Biol. 117, 227-235.
Steinmetz, M. O., Stoffier, D., Muller, S. A., Jahn, W., Wolpensinger, B., Goldie, K. N., Engel, A., Faulstich, H., and Aebi, U. (1998). Evaluating atomic models of F-actin with an undecagold-tagged phalloidin derivative. J. Mol. Biol. 276, 1-6.
Slot, J. W., and Geuze, H., J. (1985). A new method of preparing gold probes for multilabelling cytochemistry. Eur. J. Cell Biol. 38, 87-93.
Zlotnick, A., Cheng, N., Stahl, S. J., Conway, J. F. Steven, A. C., and Wingfield, P. T. (1997). Localisation of the C-terminus of the assembly domain of hepatitis B virus capsid protein: Implications for morphogenesis and organisation of encapsidated RNA. Proc. Natl. Acad. Sci. USA 94, 9556-9561.
Support our developers




