Dissecting Pathways; in Situ Electroporation for the Study of Signal Transduction and Gap Junctional Communication
Electroporation has been used for the introduction of DNA and proteins, as well as various nonpermeant drugs and metabolites into cultured mammalian cells (reviewed in Neumann et al., 2000; Chang et al., 1992). Most electroporation techniques for adherent cells involve the delivery of the electrical pulse while the cells are in suspension. However, the detachment of these cells from their substratum by trypsin or EDTA can cause significant metabolic alterations (Matsumura et al., 1982), while the efficient incorporation of proteins, peptides, or drugs without cellular damage is an especially crucial requirement, since contrary to DNA, no convenient large-scale method exists for the selection of viable from damaged cells or cells where no introduction took place after electroporation, for most proteins or drugs of interest. For these reasons, a number of approaches have been taken to bypass this problem (Kwee et al., 1990; Yang et al., 1995).
The purity of the material to be electroporated is of paramount importance. Substances such as detergents, preservatives, or antibiotics could kill the cells into which they are electroporated, even if they have no deleterious effects if added to the culture medium of nonelectroporated cells.
Dulbecco's modification of Eagle's medium (DMEM) is from ICN (Cat. No. 10-331-22). Fetal calf serum (Cat. No. 2406000AJ) and phosphate-buffered saline (PBS, Cat. No. 20012-027) are from Gibco Life Technologies. Calf serum is from ICN (Cat. No. 29-131- 54). EGF is from Intergen (Cat. No. 4110-80). HEPES (Cat. No. H-9136), Lucifer yellow CH dilithium salt (Cat. No. L-0259), trypsin (Cat. No. T-0646), MgCl2 (Cat. No. M-8266), Triton (Cat. No. T-6878), deoxycholate (Cat. No. D-5760), EDTA (Cat. No. E-5134), EGTA (Cat. No. E-3889), phenylmethylsulfonyl fluoride (PMSF, P-7626), aprotinin (Cat. No. A-6279), leupeptin (Cat. No. L-2023), benzamidine (Cat. No. B- 6506), dithiothreitol (DTT, Cat. No. D-9779), CaCl2 (Cat. No. C-4901), Tris (Cat. No. T-6791), paraformaldehyde (Cat. No. P-6148), bovine serum albumin (BSA, Cat. No. A-4503), SigmaFast DAB kit (Cat. No. D-9167 and Cat. No. U-5005), and sodium orthovanadate (Cat. No. S-6508) are from Sigma. Extran-300 detergent (Cat. No. B80002), SDS (Cat. No. 44244), NP-40 (Cat. No. 56009), glycerol (Cat. No. B10118), and peroxide (B 80017) are from BDH. CelTakTM is from BD Biosciences (Cat. No. 354240). The cell staining kit, including goat serum, secondary antibody, and avidin-biotin complex, was from Vector Labs (Vectastain kit Cat. No. PK-6101). Rabbit antipeptide antibodies against the double threonine and tyrosine phosphorylated (activated) Erk½ kinase are from Biosource International (Cat. No. 44-680). When stored frozen in aliquots, they are stable for more than 5 years. They are used at 1:500 for immunostaining and 1:10,000 for Western blotting. Three- and 6-cm tissue culture dishes are from Corning or Sarstedt. Please note that only these brands of plates fit the electroporation stand.
The system for electroporation in situ (Epizap Model EZ-16) can be purchased from Ask Science Products Inc. (Kingston, Ontario, Canada, phone: 613 545-3794). The inverted, phase-contrast and fluorescence microscope, equipped with filters for Lucifer yellow and fluorescein, was from Olympus (Model IX70).
The technique of in situ electroporation can be used equally effectively for large-scale biochemical experiments (Giorgetti-Peraldi et al., 1997; Boccaccio et al., 1998; Bardelli et al., 1998) or for the detection of biochemical or morphological changes in situ (Raptis et al., 2000a). Cells are grown on glass slides coated with conductive and transparent indium-tin oxide (ITO). The cell growth area is defined by a "window" formed with an electrically insulating frame made of Teflon as shown. A stainless-steel electrode is placed on top of the cells resting on the frame and an electrical pulse of the appropriate strength is applied, as illustrated in Fig. 1 (see also chapter 43 by Raptis et al.). The technique can be applied to a large variety of adherent cell types (Brownell et al., 1996). Cells that do not adhere well can be grown and electroporated on the same conductive slides coated with CelTakTM or poly-L-lysine used according to the manufacturer's instructions.
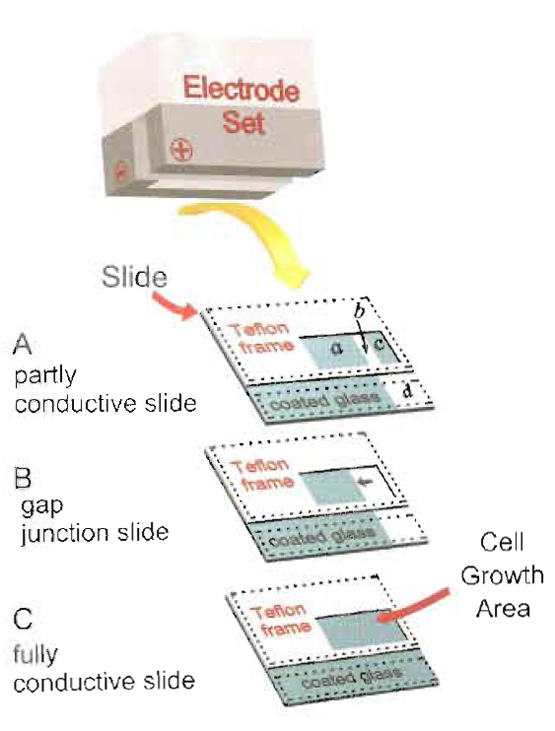 |
| FIGURE 1 Electroporation electrode and slide assembly. Cells are grown on glass slides coated with conductive and transparent ITO within a "window" cut into a Teflon frame as shown. The window can be different sizes, depending on the cell growth area required. The peptide solution is added to the cells and introduced by an electrical pulse delivered through the electrode set, which is placed directly on the frame. Dotted lines point to the positions of negative and positive electrodes during the pulse. Three slide configurations are described. (A) Partly conductive slide assembly, with electroporated (a) and non-electroporated (c) cells growing on the same type of ITO-coated surface. (b) area where the conductive coating has been stripped, exposing the non-conductive glass underneath. Cells growing in areas b and c are not electroporated (Fig. 3). (B) Partly conductive slide assembly for use in the examination of gap junctional, intercellular communication. Arrow points to the transition line between conductive and non-conductive areas (Fig. 4). (C) Fully conductive slide assembly for use in biochemical experiments. In the setup shown, cell growth area can be up to 7 × 15 mm, but larger slides and electrodes offer larger areas, up to 32 × 10mm (Fig. 2). |
A. Electroporation of Peptides into Large Numbers of Cells for Large-Scale Biochemical Experiments. Use of Fully Conductive Slides
To study the effect of protein interactions in vivo on cellular functions, such complexes can be disrupted through the introduction of peptides corresponding to the proteins' point(s) of contact. An example of this approach is described here.
Spent medium: Grow cells to confluence in DMEM with 10% calf serum. Seven days postconfluence, collect the culture supernatant and dilute 1:1 with fresh DMEM. Growth-arrest cells by incubating in spent medium prepared from the same line.
Lucifer yellow solution, 5mg/ml: To make 10ml, dissolve 50mg Lucifer yellow in 10ml calcium-free DMEM. Stable at 4°C for at least a month.
Peptides: The peptide concentration required varies with the strength of the signal to be inhibited. For the inhibition of the EGF-mediated Erk activation, prepare a solution of 5-10mg/ml (~5-10mM) of the Grb2-SH2-blocking peptide (PVPE-pmp-INQS, MW 1123 Da) in calcium-free DMEM (see Comment 1).
Epidermal growth factor: To make a 10,000× stock solution, dissolve 100 µg of lyophilised EGF in 100 µl sterile water and freeze in 5µl aliquots. Just prior to the experiment, add 1µl stock solution to 10ml calcium-free DMEM (final concentration, 100ng/ ml). The stock solution is stable at -20° or -70°C for up to 2 months.
Lysis buffer: 50mM HEPES, pH 7.4, 150mM NaCl, 10mM EDTA, 10mM Na4P2O7, 100mm NaF, 2mM vanadate, 0.5 mM PMSF, 10µg/ml aprotinin, 10µg/ml leupeptin, 1% Triton X-100.
- Choice of slides. For Western blotting experiments on cell extracts following electroporation, use fully conductive slides (Fig. 1C). Since the custommade peptide is usually the most expensive reagent in this application, to avoid waste, choose the smallest possible cell growth area which provides a sufficient number of cells. Cell growth areas of 32 × 10mm are generally sufficient to detect Erk½ activity inhibition by the Grb2-SH2 blocking peptide in EGF-stimulated, mouse NIH3T3 fibroblasts. In this case, the volume of the solution under the electrode is ~140µl and will contain approximately 700-1,400µg peptide in calcium-free DMEM. If fewer cells suffice, then slides with a cell growth area of 7 × 15mm can be used, requiring ~40µl of peptide solution. However, for the determination of [3H]thymidine uptake, cell growth areas of 7 × 4mm are preferred and they require only ~14µl of solution (see Comment 2).
- Plate the cells. Uniform spreading of the cells is very important, as the optimal voltage depends in part on the degree of cell contact with the conductive surface (see Comment 3). Add a sufficient amount of medium (DMEM containing 10% calf serum) to cover the slide (approximately 9 ml for a 6 cm dish). Pipette the cell suspension in the window cut in the Teflon frame (Fig. 1) and place the petris in a tissue-culture incubator until confluent.
- Prior to the experiment, starve the cells overnight in DMEM without serum. Alternatively, cells can be incubated in spent medium for 48h; this treatment offers wider margins of voltage tolerance (see Comment 3).
- Prior to pulse application, remove the growth medium and wash the cells gently once with calciumfree DMEM.
- Carefully wipe the Teflon frame with a folded Kleenex tissue to create a dry area on which a meniscus can form (see Pitfall 1).
- Add the peptide solution to the cells with a micropipettor in calcium-free DMEM.
- Carefully place the electrode on top of the cells and clamp it in place. To ensure electrical contact, a sufficient amount of growth medium or PBS should be present under the positive contact bar. Make sure there are no air bubbles under the negative electrode. If necessary, the electrode can be sterilized with 80% ethanol before the pulse and the procedure carried out in a laminar flow hood, using sterile solutions.
- Apply three to six pulses of the appropriate voltage and capacitance (see Comment 3).
- Remove the electrode set. Since usually only a small fraction of the material enters the cells, the peptide solution may be carefully aspirated and used again. However, care must be exercised so that the cells do not dry (see Pitfall 1).
- Add serum-free growth medium and incubate the cells for 2-5 min at 37°C to recover.
- Add EGF to the medium to a final concentration of 100ng/ml for 5min. Controls receive the same volume of calcium-free DMEM.
- Extract the cells with 50µl extraction buffer for a cell growth area of 32 × 10mm. For smaller cell growth areas, the voltage can be adjusted accordingly.
- To detect activated Erk½, load 100µg of total cell extract protein on an acrylamide-SDS gel and analyse by Western immunoblotting using the antibody directed against the dually phosphorylated, i.e., activated, form of Erk½.
- For examination of the effect of the peptide upon [3H]thymidine incorporation into DNA, serum-starve 50% confluent, NIH3T3 cells as described earlier and electroporate in the presence or absence of peptide. Incubate in medium with or without EGF for 12h at 37°C, followed by a 2h incubation with 50µCi/ml [3H]thymidine. Wash the cells with PBS and measure acid-precipitable counts. Growth areas of 4 × 7 mm are sufficient for this experiment, and [3H]thymidine can be added to the window only, in a volume of ~50µl which is held in place by surface tension.
![FIGURE 2 (A) The Grb2-SH2 blocking peptide inhibits EGF-mediated Erk activation in living cells; detection by Western blotting. The Grb2-SH2 blocking peptide was electroporated into NIH3T3 cells growing on fully conductive slides (inset, Fig. 1C, cell growth area 32 × 10mm) and growth-arrested by serum starvation. After a 5min incubation in DMEM, cells were stimulated with 100 ng/ml EGF (lanes 2-4) for 5 min. Proteins in detergent cell lysates were resolved by polyacrylamide gel electrophoresis and analysed by Western blotting using the antibody against the dually phosphorylated, active Erk enzymes. Lane 1, control, unstimulated cells; lane 2, control non-electroporated, EGF-treated cells; lane 3, cells electroporated with the control, phenylalanine-containing peptide and EGF stimulated; and lane 4, cells electroporated with the Grb2-SH2-binding peptide and EGF stimulated. From Raptis et al. (2000), reprinted with permission. (B) The Grb2-SH2 blocking peptide inhibits EGF-mediated DNA synthesis. The Grb2-SH2 blocking peptide (Pmp) or its phenylalaline-containing counterpart (phe) were electroporated at the indicated concentrations into NIH3T3 cells growing on fully conductive slides (Fig. 1C, cell growth area, 4 × 7mm) and growth-arrested by serum starvation. Following incubation at 37°C and stimulation with EGF or 10% calf serum for 12h, cells were labelled for 2h with 50µCi/ml [3H]thymidine and acid-precipitable radioactivity was determined. Numbers represent the mean ± SE from three experiments. From Raptis et al., (2000), reprinted with permission.](images/v2_pb_s09_c44_f02.jpg) |
| FIGURE 2 (A) The Grb2-SH2 blocking peptide inhibits EGF-mediated Erk activation in living cells; detection by Western blotting. The Grb2-SH2 blocking peptide was electroporated into NIH3T3 cells growing on fully conductive slides (inset, Fig. 1C, cell growth area 32 × 10mm) and growth-arrested by serum starvation. After a 5min incubation in DMEM, cells were stimulated with 100 ng/ml EGF (lanes 2-4) for 5 min. Proteins in detergent cell lysates were resolved by polyacrylamide gel electrophoresis and analysed by Western blotting using the antibody against the dually phosphorylated, active Erk enzymes. Lane 1, control, unstimulated cells; lane 2, control non-electroporated, EGF-treated cells; lane 3, cells electroporated with the control, phenylalanine-containing peptide and EGF stimulated; and lane 4, cells electroporated with the Grb2-SH2-binding peptide and EGF stimulated. From Raptis et al. (2000), reprinted with permission. (B) The Grb2-SH2 blocking peptide inhibits EGF-mediated DNA synthesis. The Grb2-SH2 blocking peptide (Pmp) or its phenylalaline-containing counterpart (phe) were electroporated at the indicated concentrations into NIH3T3 cells growing on fully conductive slides (Fig. 1C, cell growth area, 4 × 7mm) and growth-arrested by serum starvation. Following incubation at 37°C and stimulation with EGF or 10% calf serum for 12h, cells were labelled for 2h with 50µCi/ml [3H]thymidine and acid-precipitable radioactivity was determined. Numbers represent the mean ± SE from three experiments. From Raptis et al., (2000), reprinted with permission. |
Assessment of Erk activity by Western blotting following electroporation of the Grb2-SH2 blocking peptide can reveal the involvement of this domain in growth factor-mediated Erk activation. However, to ensure that the treatment itself does not cause cell stress, examination of cellular morphology, in conjunction with measurement of gene product activity by immunocytochemistry, offers a distinct advantage. This approach can demonstrate the specificity of action of the Grb2-SH2 binding peptide, as well as examine the distribution of signal inhibition across the cell layer. An added advantage is that it requires a small number of cells, hence a substantially smaller volume of the peptides (~14µl in the setup shown in Fig. 1A), compared to Western blotting (~140µl for a cell growth area of 32 × 10mm), which could be a significant consideration given their production costs. To precisely assess small background changes in morphology or gene expression levels, the presence of non-electroporated cells side by side with electroporated ones can offer a valuable control and this can be achieved by growing the cells on a conductive slide where part of the coating has been stripped by etching with acids, thus exposing the non-conductive glass underneath. However, as shown in Fig. 3, area a vs b, the slight tinge of the glass combined with the more effective staining of cells growing on ITO (possibly due to a chemical attraction of different reagents to the coating) can create problems in the interpretation of results. In addition, it was found that a number of cell lines grow slightly better on the conductive, ITO-coated glass than the nonconductive area, possibly due to the fact that the ITO-coated surface is not as smooth as glass, thus providing a better anchorage for the growth of adherent cells (Folkman and Moscona, 1978). As a result, cell density may be higher on the conductive than the etched side, which could have important implications if cell growth effects are being studied. It follows that, to assess the effect of the peptide, it is important to compare the staining and morphology of electroporated cells with non-electroporated ones while both are growing on the same type of surface. This was achieved by plating the cells on a slide where the conductive coating was removed in the pattern shown in Fig. 1A (Firth et al., 1997). A thin line of plain glass separates the electroporated and control areas while etching extends to area Fig. 1A, d so that there is no electrical contact between the positive contact bar and area Fig. 1A,c. Application of the pulse results in electroporation of the cells growing in area Fig. 1A, a exclusively, while cells growing in area Fig. 1A,b or c do not receive any pulse. In this configuration, electroporated cells are being compared to nonelectroporated ones, while both are growing on ITO-coated glass. Because the coating is only ~1,600Å thick, this transition zone does not alter the growth of cells across it and is clearly visible microscopically, even under a cell monolayer (Figs. 3 and 4).
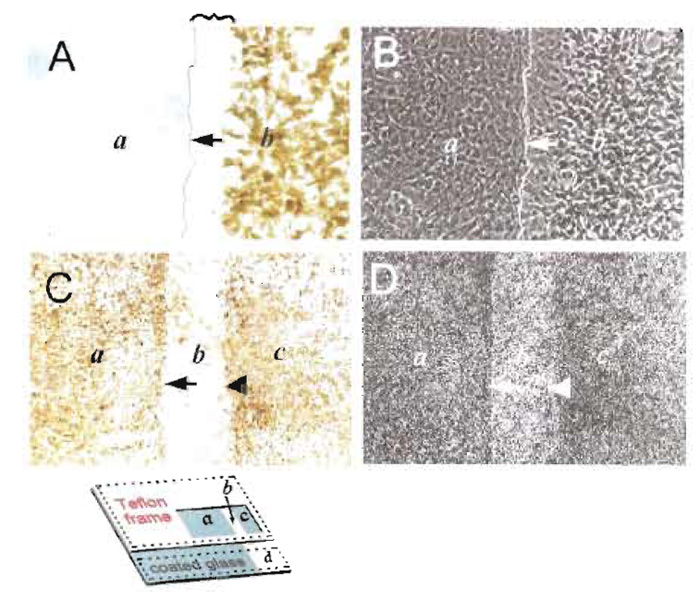 |
| FIGURE 3 The Grb2-SH2 blocking peptide inhibits EGF-mediated Erk activation in living cells; detection by immunocytochemistry. The Grb2-SH2 blocking peptide (A and B) or its control, phenylalaninecontaining counterpart (C and D) were introduced by in situ electroporation into NIH3T3 cells growing on partly conductive slides (inset, Fig. 1A) and growth arrested in spent medium. Five minutes after pulse application, cells were stimulated with EGF for 5 rain, fixed, and probed for activated Erk½, and cells from the same field were photographed under bright-field (A and C) or phase-contrast (B and D) illumination. Magnification: A and B, 240×, C and D, 40×. Arrow points to the transition line between stripped (b) and electroporated (a) areas, while arrowhead points to the line between control ITO-coated (c) and stripped (b) areas (Fig. 1A). Cells growing on the left side (a) are electroporated, whereas cells on the stripped zone (b) or right side (c) of the slide do not receive any pulse. Note that the Grb2-SH2 blocking peptide dramatically reduced the EGF signal (A, a), whereas the degree of Erk activation is the same on both sides of the slide (a or c) for cells electroporated with the control, phenylalanine-containing peptide (C). In A, inhibition of the signal extends into approximatily three to four rows of adjacent cells in the non-electroporated area (squiggly bracket in b), probably due to movement of the peptide through gap junctions (Raptis et al., 1994). At the same time, there is no detectable effect on cell morphology as shown by phase contrast (B and D). From Rapfis et al. (2000), reprinted with permission. |
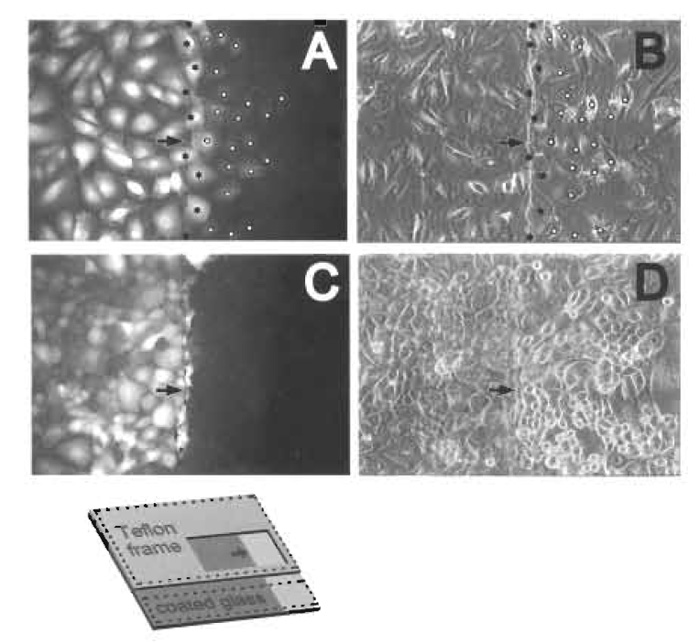 |
| FIGURE 4 In situ electroporation on a partly conductive slide for the measurement of intercellular, junctional communication. (A and B) An established, mouse lung epithelial type II line (El0) was plated on partly conductive slides (inset, Fig. 1B) and at confluence was electroporated in the presence of 5mg/ml Lucifer yellow. After washing away any unincorporated dye, cells from the same field were photographed under fluorescence (A) or phase-contrast (B) illumination (Raptis et al., 1994). Note the gradient of fluorescence indicating dye transfer through gap junctions. To quantitate intercellular communication, the number of cells into which the dye transferred through gap junctions per electroporated border cell was calculated by dividing the total number of fluorescing cells on the non-conductive side (white circles) by the number of cells growing at the border with the conductive coating (black stars). (C and D) A spontaneous transformant of the El0 line (line E9), was plated on partly conductive slides, electroporated, washed, and photographed as described earlier. Fluorescence (C) and phase-contrast (D) illumination photograph of the same field. Note the absence of dye transfer through gap junctions. In all photographs, the left side is conductive. Arrows on the conductive side point to the interphase between conductive and non-conductive areas. Magnification: 200×. From Vultur et al. (2003), reprinted with permission. |
Solutions
Peptide solution: 5-10mg/ml in calcium-free DMEM. See Section III,A and Comment 1
Lucifer yellow solution: 5 mg/ml in calcium-free DMEM. See Section III,A
Epidermal growth factor: 100ng/ml in calcium-free DMEM. See Section III,A.
- Choice of slides. Use partly conductive slides where the coating has been removed in a line as shown in Fig. 1A.
- Plate the cells as described earlier and starve them from serum.
- Aspirate the medium and wash the cells once with calcium-free DMEM.
- Add the peptide solution as described previously.
- Apply a pulse of the appropriate strength (see Comment 3).
- Add serum-free growth medium and place the cells in a 37°C incubator for the pores to close (2-5 min).
- Treat the cells with EGF for 5 min as in Section III,A.
- Fix the cells with 4% paraformaldehyde and probe with the anti-active Erk antibody according to the manufacturer's instructions.
The introduction of peptides to interrupt signaling pathways using the modification of in situ electroporation described is a powerful approach for the in vivo assessment of the relevance of in vitro interactions. Results presented in Fig. 2 and 3 clearly demonstrate that an essentially complete and specific inhibition of EGF-dependent Erk activation can be achieved through peptide electroporation. The stepwise dissection of signaling cascades is essential for the understanding of normal proliferative pathways, which could lead to the development of drugs for the rational treatment of neoplasia.
One of the targets of a variety of signals stemming from growth factors or oncogenes may be membrane channels, which serve as conduits for the passage of small molecules between the interiors of cells. Oncogene expression and neoplasia invariably result in a decrease in gap junctional, intercellular communication (GJIC) (Goodenough et al., 1996). The investigation of junctional permeability is often conducted through microinjection of a fluorescent dye such as Lucifer yellow, followed by observation of its migration into neighboring cells. This is a time-consuming approach, requiring expensive equipment, while the mechanical manipulation of the cells may disturb cellto- cell contact areas, interrupt gap junctions, and cause artefactual uncoupling. These problems can be overcome using a setup where cells are grown on a glass slide, half of which is coated with electrically conductive, optically transparent, indium-tin oxide. An electric pulse is applied in the presence of Lucifer yellow, causing its penetration into cells growing on the conductive part of the slide, and migration of the dye to non-electroporated cells growing on the nonconductive area is observed microscopically under fluorescence illumination.
The technique can be applied to a large variety of adherent cell types, including primary human lung carcinoma cells (Tomai et al., 1998; Raptis et al., 1994; Brownell et al., 1996; Vultur et al., 2003).
Solutions
Lucifer yellow solution: 5 mg/ml in calcium-free DMEM or other growth medium. See Section III.A
Calcium-free growth medium with or without 5% dialysed serum
- Plate the cells on partly conductive slides in 3 cm petris. Electroporated areas can be 4 × 4mm and non-electroporated ones 4 × 3 mm (Fig. 1B). Other slide configurations are also available (Raptis et al., 2000b).
- Aspirate the medium. Wash the cells with calcium-free DMEM.
- Add the Lucifer yellow solution.
- Apply a pulse of the appropriate strength so that cells growing on the conductive coating at the border with the non-conductive area are electroporated without being damaged. As described in Comment 3, this area receives slightly larger amounts of current than the rest of the conductive growth surface.
- Add calcium-free DMEM containing 5% dialysed serum, remove the electrode, and incubate the cells for 3-5 min in a 37°C, CO2 incubator. The inclusion of dialysed serum at this point helps pore closure.
- Wash the unincorporated dye with calcium-free growth medium.
- Microscopically examine under fluorescence and phase-contrast illumination (Fig. 4).
- Quantitate intercellular communication. Photograph the cells with a 20× objective under fluorescence and phase contrast illumination (Figs. 4A and 4B). Identify and mark electroporated cells at the border with the non-conductive area (black stars) and fluorescing cells on the nonconductive side (white circles) where the dye has transferred through gap junctions. Divide the total number of fluorescing cells on the non-conductive area by the number of electroporated cells along the border with the etched side. The transfer from at least 200 contiguous electroporated border cells is calculated for each experiment (Raptis et al., 1994). A careful kinetic analysis of dye transfer from 30s to 2h showed that the observed transfer is essentially complete by 5 min for all lines tested, while fluorescence is eliminated from the cells within approximately 60min. After the transfer is complete, cells can be fixed with formaldehyde, in which case fluorescence is retained for approximately 2 h.
1. Peptides
The concentration of peptide required varies with the strength of the signal to be inhibited. For example, for the inhibition of the HGF-mediated Stat3 activation in MDCK cells, a concentration of 1 µg/ml of a peptide blocking the SH2 domain of Stat3 (PYVNV) is sufficient (Boccaccio et al., 1998), whereas for the inhibition of the EGF-mediated Erk activation in a variety of fibroblasts or epithelial cells, a concentration of 5-10mg/ml (~5-10mM) of the Grb2-SH2 blocking peptide is necessary (Raptis et al., 2000a). The purity of the material is of utmost importance. Peptides must be HPLCpurified because impurities can cause cell death or give unexpected results. The pH of the peptide solution must be neutral, as indicated by the color of the DMEM medium where the peptide is dissolved. If it is too acidic, then it must be carefully neutralised with NaOH. In this case, the salt concentration of the nopeptide controls (DMEM without calcium) must be adjusted to the same level with NaCl because a change in conductivity may affect the optimal voltage required (see Comment 3).
Any peptide which is soluble in DMEM or other aqueous buffer can be very effectively electroporated. Good solubility is especially important because the concentration needed for effective signal inhibition can be as high as 10 mg/ml. It was nevertheless found that, at least for certain applications, the inclusion of DMSO in the electroporation solution at a concentration of up to 5%, which might aid peptide solubility, did not affect results significantly. However, a number of peptides, e.g., peptides made as fusions with the homeobox domain or other membrane translocation sequences, are usually not sufficiently soluble for this application.
As described in detail in Chapter 43 by Raptis et al., to obtain a uniform electrical field intensity over the entire area below the negative electrode, despite the fact that the conductive coating exhibits a significant amount of electrical resistance, the bottom surface of the negative electrode must be inclined relative to the glass surface in a manner proportional to the resistance of the coating. For electroporation of peptides, to minimise the volume of custom-made peptide used, slides with a conductivity of 2Ω/sq, the most conductive commercial grade available, are used. The slides and electrodes come in different sizes, with the biggest cell growth area in this configuration being 32 × 10mm. Depending on the experiment, if larger numbers of cells are required, extracts from two to three slides may be pooled. In this case, the peptide solution can be aspirated and used again. Alternatively, an electrode configuration with two positive contact bars can be employed, as described in Chapter 43 by Raptis et al.
The slides come with the apparatus, individually wrapped and sterile. However, they can be reused many times after washing with Extran-300 detergent while scrubbing with a toothbrush. In this case they must be sterilized with 80% ethanol for 20min and the ethanol rinsed with sterile distilled water prior to plating the cells. Alternatively, the slides can be gassterilized. Do not autoclave. The glass can withstand high temperatures, but autoclaving would damage the Teflon frames.
Electrical field strength has been shown to be a critical parameter for cell permeation, as well as viability (Chang et al., 1992). It is generally easier to select a discrete capacitance value for a given electroporated area and space between the conductive coating and the negative electrode and then precisely control the voltage. Both parameters depend upon the size of the electroporated area; larger conductive growth areas necessitate higher voltages and/or higher capacitances for optimal permeation. For the 32 × 10 mm cell growth area, some damage to the coating may be noted at the higher voltages necessary if a single pulse is employed. However, using higher capacitance values and multiple pulses with lower voltage settings can yield efficient cell permeation with no damage to the coating, and this treatment is also better tolerated by the cells. For greater growth areas, the dual positive contact bar design described in Chapter 43 by Raptis et al. can be employed.
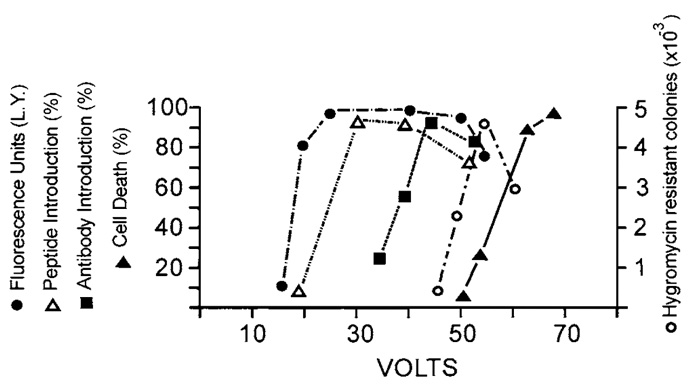 |
| FIGURE 5 Effect of field strength on the introduction of different molecules. Three pulses of increasing voltage were applied to confluent rat F111 fibroblasts growing on a conductive surface of 32 × 10mm from a 32µF capacitor in the presence of 5 mg/ml Lucifer yellow ( |
 |
| FIGURE 6 Determination of the optimal voltage. Rat F111 fibroblasts growing on partly conductive slides (Fig. 1B, conductive area, 4 × 4 mm) were electroporated in the presence of 5 mg/ml Lucifer yellow using three pulses of increasing voltage delivered from a 0.2µF capacitor. (A and B) 18 V, (C and D) 26V, (E and F) 28V, (G and H) 32 V, and (I and J) 40V. After washing of the unincorporated dye, cells were photographed under fluorescence (A, C, E, G, I) or phase-contrast (B, D, F, H and J) illumination. Straight arrow points to the interphase between conductive (right) and non-conductive (left) areas. Curved arrows in C and D point to a cell which has been killed by the pulse. Note the dark, pycnotic and prominent nucleus under phase contrast and the flat, nonrefractile appearance. Such cells do not retain any electroporated material as shown by the absence of fluorescence (C). Note that the number of such cells along the border with the non-conductive area increases with voltage. Arrowheads in E and F point to a cell that has a prominent nucleus under phase contrast (F) but has retained the dye (E). Such cells rapidly recover their normal morphology, indistinguishable from their non-electroporated counterparts. White arrowheads in I and J point to membrane blebs which tend to enclose Lucifer yellow and fluoresce strongly. Such membrane blebbing tends to be more prominent under higher voltages. Note that if the determination of intercellular communication is desired, then the voltage must be such that cells at the border with the non-conductive area are electroporated without being damaged (e.g., 18V, A and B), whereas for all other applications, voltages of approximately 26-32 V would be preferred (C to H). Magnification: A and B, 120X; C-J, 240×. |
Prepare a series of slides with cells plated uniformly in a 4 × 7mm window on a partly conductive slide (Figs. 1A or 1B). Set the apparatus at 0.5 µF capacitance. Prepare a solution of 5mg/ml Lucifer yellow and electroporate at different voltages (0.5 µF, three pulses) to determine the upper limits where a small fraction of the cells at the border with the etched side (probably the more extended ones) are killed by the pulse, as determined by visual examination under phase-contrast and fluorescence illumination (Fig. 6). Depending on the cells, this voltage can vary from 20 to 40 V. Repeat the electroporation using the peptide solution at different voltages starting at 10 V below the upper limit and at 2 V increments. The Lucifer yellow dye offers an easy way to test for cell permeation.
Under the appropriate conditions, electroporation was shown not to affect the activity of Erk½ or the stress-activated kinases JNK/SAPK and p38hog This was shown by probing with antibodies specific for the activated forms of these kinases (Fig. 7); no activation of JNK/SAPK or p38hog was found under conditions of up to 70V (Figs. 7C and 7E). These kinases were, however, slightly activated at voltages higher than 85 V, when more than 60% of the cells were killed by the pulse (not shown).
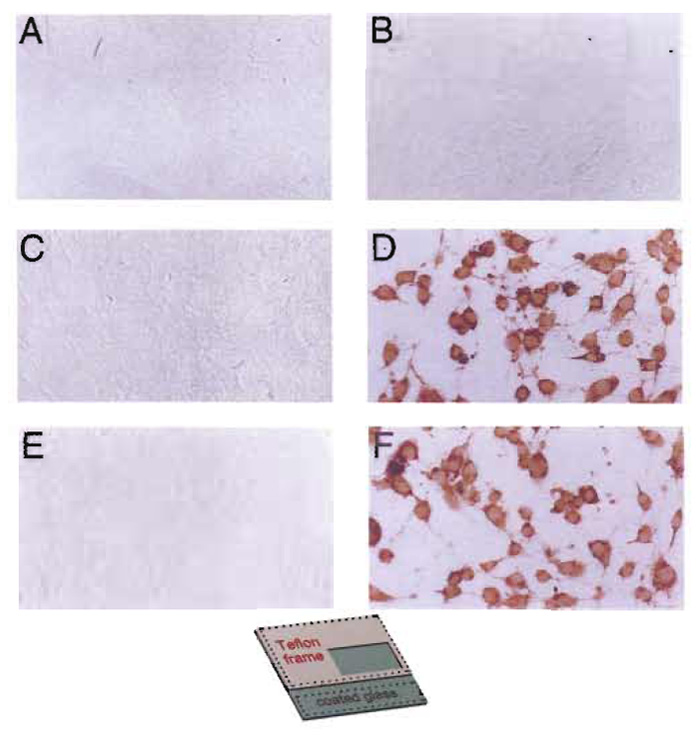 |
| FIGURE 7 In situ electroporation does not affect ERK activity or the stress pathway. (A, C, and E) NIH3T3 cells were plated on fully conductive slides (inset, Fig. 1C, conductive growth area 4 × 8mm), growth arrested in 50% spent medium, and electroporated in the presence of PBS containing 0.025% DMSO (0.2µF, 70 V, four pulses). Ten minutes after the pulse, cells were fixed and stained for activated ERK (A), activated JNK/SAPK (C), or activated p38hog (E), respectively. Electroporated cells were photographed under brightfield illumination. (B, D, and F) NIH3T3 cells were plated on conductive slides, treated with UV light for 10rain, fixed, and stained for activated ERK (B), activated JNK/SAPK (D), or activated p38hog (F), respectively. From Brownell et al. (1998), reprinted with permission. Magnification: 240×. |
- Care must be taken so that cells do not dry during the procedure, especially during wiping of the frame with a tightly folded Kleenex. It was found that serum-starved cells were more susceptible than their counterparts grown in medium containing serum. The morphology of cells that have been killed by drying is very similar to cells that have been killed by the pulse (Fig. 6). Slightly dried cells may incorporate Lucifer yellow and appear almost normal under phase contrast, whereas cells that have dried to a great extent display dark nuclei and may not retain Lucifer yellow. It was also found that the combination of even slight drying with electroporation may have undesirable effects on gene expression (e.g., induction of fos by serum; unpublished observations). In the case of electroporation on a partly conductive slide (Figs. 1A or 1B), drying of the cells is immediately suspected if cells growing on the non-electroporated area exhibit Lucifer yellow fluorescence.
- Accurate determination of the optimal voltage is very important. The limits of voltage tolerance are narrower for serum-starved cells (Brownell et al., 1997) or if the introduction of larger molecules is attempted.
- For the determination of GJIC, it is important to wash the dye using a calcium-free solution (growth medium or PBS). If calcium-containing growth medium is used instead, the values obtained may be reduced, presumably because of the calcium influx, which was shown to interrupt junctional communication.
The financial assistance of the Canadian Institutes of Health Research, the Canadian Breast Cancer Research Initiative, the Natural Sciences and Engineering Research Council of Canada (NSERC), and the Cancer Research Society Inc. is gratefully acknowledged. AV is the recipient of NSERC and Ontario Graduate studentships, a Queen's University graduate award, and a Queen's University travel grant. HB was the recipient of a studentship from the Medical Research Council of Canada and a Microbix Inc. travel award. We are grateful to Dr. Erik Schaefer of Biosource Int. for numerous suggestions and valuable discussions.
References
Bardelli, A., Longati, P., Gramaglia, D., Basilico, C., Tamagnone, L., Giordano, S., Ballinari, D., Michieli, P., and Comoglio, P. M. (1998). Uncoupling signal transducers from oncogenic MET mutants abrogates cell transformation and inhibits invasive growth. Proc. Natl. Acad. Sci. USA 95, 14379-14383.
Boccaccio, C., Ando, M., Tamagnone, L., Bardelli, A., Michielli, P., Battistini, C., and Comoglio, P. M. (1998). Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 391, 285-288.
Brownell, H. L., Firth, K. L., Kawauchi, K., Delovitch, T. L., and Raptis, L. (1997). A novel technique for the study of Ras activation; electroporation of [α32P]GTP. DNA Cell Biol. 16, 103-110.
Brownell, H. L., Lydon, N., Schaefer, E., Roberts, T. M., and Raptis, L. (1998). Inhibition of epidermal growth factor-mediated ERK½ activation by in situ electroporation of nonpermeant [(alkylamino)methyl]acrylophenone derivatives. DNA Cell Biol. 17, 265-274.
Brownell, H. L., Narsimhan, R., Corbley, M. J., Mann, V. M., Whitfield, J. F., and Raptis, L. (1996). Ras is involved in gap junction closure in mouse fibroblasts or preadipocytes but not in differentiated adipocytes. DNA Cell Biol. 15, 443-451.
Chang, D. C., Chassy, B. M., Saunders, J. A., and Sowers, A. E. (1992). "Guide to Electroporation and Electrofusion." Academic Press, New York.
Firth, K. L., Brownell, H. L., and Raptis, L. (1997). Improved procedure for electroporation of peptides into adherent cells in situ. Biotechniques 23, 644-645.
Gambarotta, G., Boccaccio, C., Giordano, S., Ando, M., Stella, M. C., and Comoglio, P. M. (1996). Ets up-regulates met transcription. Oncogene 13, 1911-1917.
Giorgetti-Peraldi, S., Ottinger, E., Wolf, G., Ye, B., Burke, T. R., and Shoelson, S. E. (1997). Cellular effects of phosphotyrosinebinding domain inhibitors on insulin receptor signalling and trafficking. Mol. Cell. Biol. 17, 1180-1188.
Goodenough, D. A., Goliger, J. A., and Paul, D. L. (1996). Connexins, connexons, and intercellular communication. Annu. Rev. Biochem. 65, 475-502.
Kwee, S., Nielsen, H. V., and Celis, J. E. (1990). Electropermeabilization of human cultured cells grown in monolayers: Incorporation of monoclonal antibodies. Bioelectrochem. Bioenerg. 23, 65-80.
Marais, R., Spooner, R. A., Stribbling, S. M., Light, Y., Martin, J., and Springer, C. J. (1997). A cell surface tethered enzyme improves efficiency in gene-directed enzyme prodrug therapy. Nature Biotechnot. 15, 1373-1377.
Matsumura, T., Konishi, R., and Nagai, Y. (1982). Culture substrate dependence of mouse fibroblasts survival at 4°C. In Vitro 18, 510-514.
Nakashima, N., Rose, D., Xiao, S., Egawa, K., Martin, S., Haruta, T., Saltiel, A. R., and Olefsky, J. M. (1999). The functional role of crk II in actin cytoskeleton organization and mitogenesis. J. Biol. Chem. 274, 3001-3008.
Otaka, A., Nomizu, M., Smyth, M. S., Shoelson, S. E., Case, R. D., Burke, T. R., and Roller, P. P. (1994). Synthesis and structureactivity studies of SH2-binding peptides containing hydrolytically stable analogs of O-phosphotyrosine. In "Peptides; Chemistry, Structure and Biology" (R. S. Hodges and J. A. Smith, eds.), pp. 631-633. Escom, Leiden.
Raptis, L., Brownell, H. L., Firth, K. L., and MacKenzie, L. W. (1994). A novel technique for the study of intercellular, junctional communication; electroporation of adherent cells on a partly conductive slide. DNA Cell Biol. 13, 963-975.
Raptis, L., Brownell, H. L., Vultur, A. M., Ross, G., Tremblay, E., and Elliott, B. E. (2000a). Specific inhibition of growth factorstimulated ERK½ activation in intact cells by electroporation of a Grb2-SH2 binding peptide. Cell Growth Differ. 11, 293-303.
Raptis, L., and Firth, K. L. (1990). Electroporation of adherent cells in situ. DNA Cell Biol. 9, 615-621.
Raptis, L., Tomai, E., and Firth, K. L. (2000b). Improved procedure for examination of gap junctional, intercellular communication by in situ electroporation on a partly conductive slide. Biotechniques 29, 222-226.
Robinson, M. J., and Cobb, M. H. (1997). Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 9, 180-186.
Tomai, E., Brownell, H. L., Tufescu, T., Reid, K., Raptis, S., Campling, B. G., and Raptis, L. (1998). A functional assay for intercellular, junctional communication in cultured human lung carcinoma cells. Lab. Invest. 78, 639-640.
Vultur, A., Tomai, E., Peebles, K., Malkinson, A. M., Grammatikakis, N., Forkert, P. G., and Raptis, L. (2003). Gap junctional, intercellular communication in cells from urethane-induced tumors in A/J mice. DNA Cell Biol. 22, 33-40.
Williams, E. J., Dunican, D. J., Green, P. J., Howell, F. V., Derossi, D., Walsh, F. S., and Doherty, P. (1997). Selective inhibition of growth factor-stimulated mitogenesis by a cell-permeable Grb2-binding peptide. J. Biol. Chem. 272, 22349-22354.
Yang, T. A., Heiser, W. C., and Sedivy, J. M. (1995). Efficient in situ electroporation of mammalian cells grown on microporous membranes. Nucleic Acids. Res. 23, 2803-2810.
Support our developers




