Tracking Individual Chromosomes with Integrated Arrays of lacop Sites and GFP-laci
Repressor: Analyzing Position and Dynamics of Chromosomal Loci in Saccharomyces cerevisiae
The visualisation of specific DNA sequences in living cells, achieved through the integration of lac operator arrays (lacop) and expression of a GFP-lac repressor fusion, has provided new tools to examine how the nucleus is organised and how basic events such as sister chromatid separation occur (Straight et al., 1996; Belmont, 2001). In contrast to other methods, such as fluorescence in situ hybridisation, the lacop GFP-lac repressor (GFP-laci) technique is noninvasive and therefore interferes minimally with nuclear structure and function. In addition, it facilitates analysis of the rapid dynamics of specific DNA loci (Gasser, 2002). Although this technique has been adapted to organisms from bacteria to humans, the ease with which GFP fusions can be targeted to specific chromosomal sites depends on the ability of the organism to carry out homologous recombination. This process is very efficient in budding yeast, allowing pairs of chromosomal loci to be analysed at the same time through the use of two bacterial repressors (laci and tetR) fused to different GFP variants. Given the relatively advanced state of the art in budding yeast, this article presents protocols optimised for this organism. These provide a starting point for adapting multilocus tagging to other species. Moreover, the techniques described here for the quantitative analyses of locus dynamics are universally applicable.
Yeast minimal and rich media (SD, YPD) are described in Guthrie et al. (1991). Cells can be mounted on a depression slide (Milian SA, Cat. No. CAV-1, Fig. 2A) upon 1.4% agarose (Eurobio Cat. No. 018645) containing SD medium with 4% glucose (Fluka). Aliquots of this can be kept at 4 °C for several months. Alternatively, cells can be immobilised on a 18-mm coverslip treated with concanavalin A (Con A, Sigma, Cat. No. C-0412) in a cell observation chamber (Ludin chamber, Life Imaging Services, Fig. 2B). Con A dissolved to 1mg/ml in H2O is stable at -20°C for months. Widefield microscopy is performed on a Metamorph-driven Olympus IX 70 inverted microscope with Olympus Planapo 60×/NA = 1.4 or Zeiss Planapo 100×/NA = 1.4 objectives on a piezoelectric translator (PIFOC; Physik Instrumente), illuminating with a PolychromeII monochromator (T.I.L.L. Photonics). Also needed is a CoolSNAP-HQ digital camera (Roper Scientific) or equivalent, and both the FITC filter set for detecting GFP (Chroma, Ref. 41001) and the CFP/YFP filter set (e.g., Chroma, Ref. 51017). Confocal microscopy can be performed on a Zeiss LSM510 Axiovert 200M, equipped with a Zeiss Plan-Apochromat 100×/NA = 1.4 oil immersion or a Plan-Fluar 100×/NA = 1.45 oil immersion objective. The stage is equipped with a hyperfine motor HRZ 200. Temperature is stabilised using a temperature-regulated box surrounding the microscope (The Box, Life Imaging Services). Software used for analysis is (a) Excel (Microsoft), (b) ImageJ public domain software (Rasband), (c) Imaris v 3.3 (Bitplane), (d) Mathematica 4.1 (Wolfram Research), and (e) Metamorph v 4.6r6 (Universal Imaging Corp.).
A. Preparations
1. Plasmids and Strains
Yeast transformation and growth are as described (Guthrie et al., 1991). The lacop/GFP-laci system for site recognition exploits the high affinity and specificity of the bacterial lac repressor for its recognition sequence (lacop All procedures are performed analogously for the tetR/tetop system (Michaelis et al., 1997).
- Plasmids or integrations of repetitive arrays are difficult to propagate in both bacteria and yeast due to recombination induced excision events. To avoid this, bacteria should be grown at 30°C in a recombinationdeficient strain [STBL2 (Invitrogen Life Technologies) or SURE (Stratagene)].
- Integrate a copy of lac repressor fused in frame to sequences encoding the S65T V163A, S175G derivative of GFP and a nuclear localisation signal, e.g., pAFS144 into the yeast strain. This red-shifted GFP derivative has a higher emission intensity and longer fluorescence time than natural GFP (Straight et al., 1998). The laci later helps to stabilise the lacop array in yeast.
- Insert a multimerised lacop array (usually 256 copies or ~10kb) into the chromosome by standard transformation using a linearised construct that integrates by homologous recombination. Integration is directed to a genomic locus by a unique cleavage within a polymerase chain reaction (PCR)-generated genomic sequence >200bp inserted into the host plasmid (e.g., pAFS52 integration is selected by growth on SD-trp; Straight et al., 1996; Heun et al., 2001a; Hediger et al., 2002). In yeast as few as 24 contiguous lacop sites can be detected readily.
- Check the proper insertion by standard colony PCR and/or Southern blotting (Guthrie et al., 1991). Binding of laci-GFP to the lacop array results in a bright focal spot, detected readily by fluorescence microscopy within the nucleoplasm. Confirmed transformants with bright signals should be frozen and stored immediately as individual colony isolates. When strains are recovered from frozen stocks, they should be grown on selective medium to avoid further excision events.
Note: Other GFP fusions, optimised forms of CFP or YFP (or ECFP and EYFP), have also been used successfully in yeast (Lisby et al., 2003). The lac repressor used is also modified to prevent tetramerisation, thus minimising artefactual higher order interactions between lacop sites (Straight et al., 1996). - Double tagging. If the position or mobility of two genomic loci is to be compared, one should avoid tagging both with the same repeats. It has been shown that identical arrays can undergo a pairing event that, at least in the case of the tet system, depends on the expression of the repressor (tetR; Fuchs et al., 2002). By using tetop for one site, and lacop for the second, the risk of spurious pairing is eliminated. Useful pairs of GFP derivatives are CFP and YFP, or GFP and the new monomeric mRFP (Campbell et al., 2002).
- In contrast to the laci-GFP fusion (Figs. 1A and 1B), the tetR-GFP gives a high and generally diffuse nucleoplasmic background in yeast, both in the presence and in the absence of tetop repeats (Figs. 1C and 1D).
- Dynamics. If movement analysis is to be pursued, it is important to differentiate the movement of the nucleus itself or that induced by mechanical vibrations from the dynamics of the chromosome. Nuclear movement must be determined and then subtracted from that of a specifically tagged site, using any of the following methods.
- Visualisation of the nuclear envelope with Nup49-GFP (Belgareh et al., 1997; Heun et al., 2001a). In this case the nuclear centre can be interpolated from the oval or circular pore signal in an automated fashion by software such as ImageJ or Metamorph (Figs. 1A and 1B). The DNA locus position is then determined relative to the nuclear centre for each frame.
- Diffuse nucleoplasmic signal of tetR-GFP (Figs. 1C and 1D). The centre of the nucleus is defined by interpolation frame by frame and locus movement is calculated relative to this.
- By comparing the motion of two tagged loci, one can calculate average movement without concern for nuclear drift. The fact that both loci are moving has to be taken into account for movement quantitation (see later).
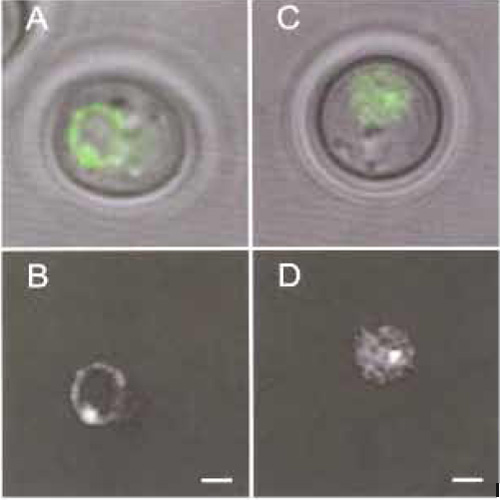 |
| FIGURE 1 (A and C) An overlay of the phase image and the fluorescence image of a GFP-tagged yeast cell in G1 phase. (B and D) The corresponding fluorescence image. The lacop array is integrated at the LYS2 locus; the nucleus is visualised by the tagged nuclear pore component Nup49-GFP (A,B) or by using the diffuse staining of nucleoplasm by tetR-GFP (C,D). Bar: 1 µm. |
- All yeast strains to be analysed should be cultured identically and preferably to an early exponential phase of growth (<0.5 × 107 cells/ml) in synthetic or YPD medium, starting from a fresh overnight culture. Depletion of glucose or growth on alternative carbon sources can alter chronetin dynamics. Wash cells once before observation to avoid YPD autofluorescence. We recommend two mounting techniques for living cell visualisation.
-
- SD-agarose-filled slides (Fig. 2A): Immobilised cells between an agarose patch on a depression slide and a coverslip to avoid flattening or distortion of the yeast by coverslip pressure on a normal glass slide. Cells sealed in this way are in a closed environment in which the depletion of O2 and production of CO2 bubbles can influence growth and impair visualization. Optimally this technique is used for imaging periods limited to <60min.
- Melt an aliquot of SD/agarose at 95°C until the agarose has completely melted, but not longer.
- Vortex briefly and transfer 150µl into the well of a depression slide that is preheated either by a heating block or by passage through the flame of a Bunsen burner.
- Immediately pass a normal microscope slide over the depression to remove excess agarose as depicted in Fig. 2A.
- While the agarose solidifies, recover the cells from 1 ml of culture by centrifugation for i min at <10,000 g.
- Resuspend the cells in ~20 µl of appropriate medium.
- Once the agarose has solidified, remove the upper slide by sliding along the depression slide surface and place ~5µl of concentrated cells on the agarose patch.
- Close with a coverslip, eliminate eventual air bubbles, and seal with nail polish.
Note: Monitor bud emergence and cell division carefully, as some brands of nail polish contain solvents that influence yeast cell physiology negatively.
- Cell observation chamber (Ludin chamber, Fig. 2B): The second technique uses a Ludin chamber in which cells are attached noncovalently to a coverslip by a lectin. The medium-filled chamber is assembled as shown in Fig. 2B. A flow of fresh medium can be applied.
- Coat 18-mm coverslips with 10µl Con A (1mg/ml in H2O) and let them air dry for >20min. Coated slides can be kept for weeks at room temperature.
- Adhere cells to the Con A-coated coverslip by sedimenting 1 ml of the culture at 1g for 3 min at room temperature.
- Remove excess culture and add ~1ml fresh preheated medium before closing the chamber.
- SD-agarose-filled slides (Fig. 2A): Immobilised cells between an agarose patch on a depression slide and a coverslip to avoid flattening or distortion of the yeast by coverslip pressure on a normal glass slide. Cells sealed in this way are in a closed environment in which the depletion of O2 and production of CO2 bubbles can influence growth and impair visualization. Optimally this technique is used for imaging periods limited to <60min.
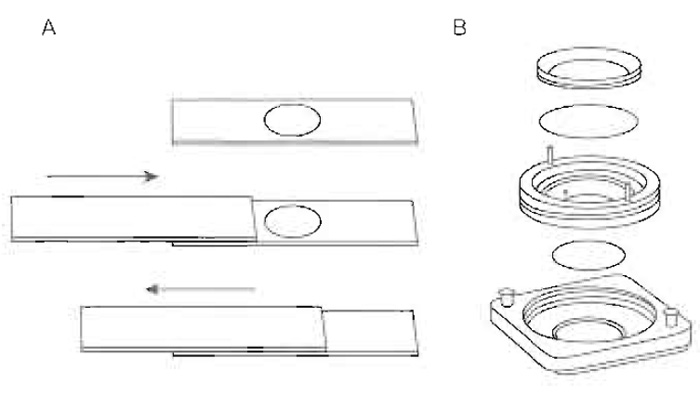 |
| FIGURE 2 Yeast cells can be immobilised for imaging either using an agarose patch on a depression slide (A) or using a cell observation chamber (e.g., Ludin chamber; B). |
In order to have a stable condition for microscopic observation, the temperature of the microscope and room should be controlled carefully (±2°C). Two mechanisms are used standardly. The first is to enclose the entire imaging part of the microscope in a commercially available temperature-regulated box (e.g., Life Imaging Services or Zeiss). A second, less precise method is to regulate the temperature of the slide through a heated stage.
B. Image Acquisition
1. General
The choice of imaging technique depends on the question being asked. To derive quantitative information on the position of a given locus relative to a fixed structure (e.g., the spindle pole body, nucleolus, or nuclear envelope), three-dimensional (3D) stacks and detection of different wavelengths may be necessary. An analysis of fine movement and chromatin dynamics, however, requires the rapid and extended capture of one or more fluorochromes. Bleaching of the signal is often a major limiting factor in time-lapse imaging. One should note that chromatin movement is very fast [movements >0.5µm in less than 10s (Heun et al., 2001a)], making it necessary to have rapid image acquisition with a minimal interval between sequential images. To optimise acquisition, parameters such as image resolution, the number of z frames, intervals between frames, light intensity, and exposure time can be varied. In all cases, it is of utmost importance to minimise and monitor laser- or light-induced damage to the organism during imaging, in part by comparing the time required for one division cycle in imaged and nonimaged cells.
Cell Cycle Determination
As position and mobility of a chromosomal locus can vary with stages of the cell cycle, it is crucial to determine precisely what stage each imaged cell is in. This is done by monitoring bud presence and bud size, as well as the shape and position of the nucleus, as visualised by the Nup49-GFP fusion and a transmission or phase image. Figure 3 summarises the morphologies that characterise each stage of the cell cycle.
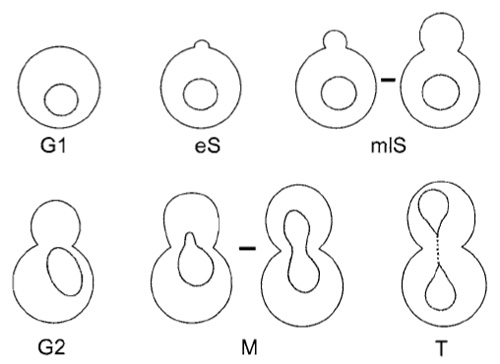 |
| FIGURE 3 Diagrams of a budding yeast cell at different characteristic points in the cell division cycle. The following criteria are used to identify the indicated stage. G1 phase, unbudded cells with round nuclei or attached pairs of posttelophase cells that have two round, clearly separated nuclei; early S, with initial bud emergence, cells are in early S; mid-to-late S, cells with a bud big enough to form a ring at the bud neck, in which nuclei are still round and centred in the mother cell; G2 phase, large budded cells (bud≥ two-thirds of mother) with the nucleus at the bud neck; mitosis (M), large budded cells in which the nucleus extends into the daughter cell due to spindle extension; telophase (T), two globular cells with two distinct nuclei that remain connected by residual NE structures. |
For the imaging of large fields of cells, best results are obtained with a wide-field microscope equipped with a PIFOC, Xenon light source, and monochromator that allows a broad and continuous range of incident light wavelengths, as well as rapid switching between these values. Images are acquired by a highspeed monochrome CCD camera run by a rapid imaging software, such as Metamorph. The limiting step is often the speed of signal transfer from the CCD chip to the RAM and/or hard disk of your computer.
z-Stacks
Wide-field microscopy is well adapted to experiments in which a large number of cells (200-300) need to be scored, e.g., when determining the subnuclear position of a given locus relative to the nuclear envelope or another tagged locus or landmark (e.g., spindle pole body or nucleolus). The reference point should optimally be tagged with a different fluorescent protein. If two loci bind the same fluorescent fusion proteins, then their intensities should be significantly different. Rapid through-focus stacks of images using the full chip capacity of the camera are taken of cells growing on agar or in a Ludin chamber (such that 20-30 individual cells are resolved per field). Optimal parameters for GFP are as follows: exposure time, 100-200ms; z spacing of 200nm for 18 focal planes, excitation wavelength 475 nm. For dual-wavelength capture, images of both wavelengths (CFP: 432nm, ~300ms; YFP: 514nm, ~150ms) must be acquired before the focal plane changes. A phase image is taken after every stack of fluorescence images. Wide-field images have out-offocus haze and deconvolution of the z stack is often necessary to reassign blurred intensities back to their original source. Use Metamorph software or other available deconvolution packages.
The conditions for capturing 3D time-lapse series are as follows: 5-11 optical z slices taken every 1 to 4min, z sections are 200 to 400nm in depth, and the exposure time is ~50 ms. Using these settings, up to 300 stacks of five sections each (1500 frames) at 1-min intervals can be captured without affecting cell cycle progression. More rapid sampling with this system, however, leads to bleaching and potential cellular damage. Until this can be remedied by more rapid and more sensitive CCD cameras, wide-field microscopy is recommended for less rapid time-lapse imaging (intervals ≥60s) on larger fields and confocal microscopy (see later) for very rapid time-lapse imaging (intervals ≤2s) on small regions of interest (typically one yeast nucleus).
For very long imaging times (>1h), stray light should be suppressed by inserting an additional shutter. Deconvolution is performed using the Metamorph fast algorithm with five iterations, a sigma parameter of 0.7, and a frequency of 4.
3. Confocal Microscopy
To follow chromatin dynamics in individual cells with rapid time-lapse microscopy, the Zeiss LSM510 scanning confocal microscope is particularly well adapted, although the laser and acousto-optic tuneable filter (AOTF) system is limited in activation wavelengths. Its positive attributes are an ability to limit scanhead motion to a minimal region of interest (ROI), rapid and well-regulated scanning speeds, and the possibility to adjust pinhole aperture and laser intensities to very low levels, while maintaining maximal sensitivity.
General Settings
To reduce the risk of damage by illumination, the laser transmission is kept as low as possible, and the cells are imaged as rapidly as possible within a minimal ROI. Useful settings for the Zeiss LSM510 are as follows.
Filters: Channel 1: Lp 505 for GFP alone; channel 1 Lp 530, channel 3 Bp 470-500 for YFP/CFP single track acquisition.
Channel setting: Pinhole 1-1.2 airy unit (corresponding to optical slice of 700 to 900nm); detector gain: 930 to 999; amplifier gain: 1-1.5; amplifier offset: 0.2-0.1 V; laser transmission AOTF = 0.1-1% for GFP alone, 1-15% for YFP, and 10-50% for CFP in single track acquisition. In order to use minimal laser transmission the pinhole must be aligned regularly.
Scan setting: Speed 10 (0.88µs/pixel); 8 bits one scan direction; 4 average/mean/line; zoom 1.8 (pixel size: 100 x 100 nm)
Imaging intervals: 1.5 s
Note: If CFP and YFP signals are very weak, images can be acquired sequentially using the more sensitive LSM 510 channel 1 in multitrack mode. This allows the use of broader filters: long-pass filter Lp 475 for CFP and Lp 530 for YFP. Alternatively, and to avoid any cross talk, recover the YFP signal as before and use Bp 470-500 on channel 3 for CFP. These latter parameters will slow the imaging process.
If maximal capture speed is desired, only one image per time point can be taken, as long as the GFP spot stays in the imaged plane of focus (called 2D time lapse). Often the plane of focus has to be changed manually to follow the spot. Image acquisition in 3D has two main advantages. (1) The GFP spot does not have to be followed manually as it is always present in one of the focal planes. A subsequent maximal projection along the z axis produces a complete 2D time sequence without loss of focus on the GFP spot. (2) After image reconstruction, one can visualise the nucleus and calculate distances in 3D. Such measurements are nonetheless compromised by the reduced optical resolution in z (≥0.5 µm for 488-nm light).
Specific 3D time-lapse settings are as follows: six to eight optical slices in z, 300- to 450-nm spacing in z with Hyperfine HRZ 200 motor using a ROI of 3 × 3 to 4 × 4 µm and time intervals of 1.5 s. A 12-min time-lapse series at 0.2% laser transmission did not influence cell cycle progression.
1. z Stacks
Determination of the subnuclear position of a GFPtagged locus is monitored relative to the centre of the Nup49-GFP ring. Nuclei in which the tagged locus is at the very top or bottom of the nucleus are not scored because the pore signal no longer forms a ring but a surface and a peripheral spot will appear internal.
- Measure the distance from the centre of intensity of the GFP spot to the nearest pore signal along the nuclear diameter, as well as the nuclear diameter itself, using the middle of the GFP-Nup49 ring as the periphery (Fig. 4). Several programs can export coordinates of points of interest, and the publicly available point picker plug in for ImageJ (Rasband) is useful.
- Calculate the distances/diameter ratio, e.g., using Excel. Determine the precise relative radial position by dividing the distance between pore and the spot by half of the calculated diameter, thus normalising distances.
- Classify the position of each spot with respect to three concentric zones of equal surface (Fig. 4). The peripheral zone (zone I) is a ring of width = 0.184× the nuclear radius (r). Zone II lies between 0.184 and 0.422r and zone III is the core of the nucleus with radius = 0.578 r. The three zones are of equal surface no matter where the nuclear cross section is taken.
- Compare the measured distribution to another (e.g., other cell cycle phase, another condition or a random distribution) with a χ2 analysis. If only percentages of one zone (e.g., the outermost zone) are compared for different conditions (or to a random distribution), a proportional test should be applied. Statistical significance is determined using a 95% confidence interval.
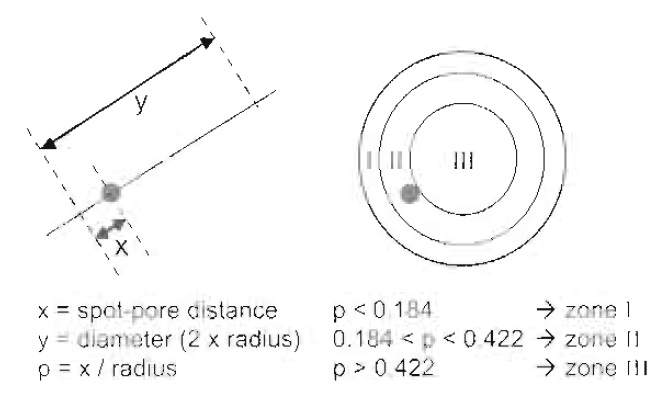 |
| FIGURE 4 Analysis of DNA locus position. Relative locus position is calculated by normalising measured distance × by the radius (0.5 × measured distance y). The relative radial distances can then be classified and attributed to three groups of equal surface. The peripheral zone (zone I) is a ring of width = 0.184 × the nuclear radius (r). Zone II lies between 0.184 and 0.422r, and zone III is the centre of the nucleus with radius = 0.578 r. In a predicted random distribution every group would contain one-third of the cells. |
Locus Tracking
A prerequisite for the precise description of chromatin movement is the knowledge of the coordinates of the locus and of the nuclear centre for each frame of a time-lapse movie. In collaboration with D. Sage and M. Unser (Swiss Federal Institute of Technology, Lausanne), a best-fit algorithm has been developed that reliably tracks a moving spot in 2D time-lapse movies or in maximal projections of z stacks in 3D time lapse using nuclei carrying Nup49-GFP or expressing tetR-GFP to detect the nucleoplasmic signal. This system is complete and dramatically improves reproducibility and the speed of analysis, while allowing user intervention at several stages. The algorithm has been implemented as a Java plug in for the public domain ImageJ software (Rasband; Sage et al., 2003). The spatiotemporal trajectory is exported as x,y coordinates for each time point in a spreadsheet. An implementation for 3D image stacks over time is also available (Sage et al, 2005). Automated image analysis requires three steps.
- Alignment phase. The first step is an alignment module that compensates for the translational movement of the nucleus, cell, or microscope stage. This is achieved by a modifiable threshold on the image. The extracted points are then fitted within an ellipse using the least-squares method. Finally, each image is realigned automatically with respect to the centre of the ellipse.
- Preprocessing phase. To facilitate the detection of the tagged locus, the images are convolved with a Mexican-hat filter. This preprocessing compensates for background variations and enhances small spot-like structures.
- Tracking phase. The final step is the tracking algorithm. Using dynamic programming, which takes advantage of the strong dependency of the spot position in one frame on its position in the next, the optimal trajectory over the entire period of the movie is determined. The following three criteria influence spot recognition: (1) maximum intensity (i.e., the tagged DNA is usually brighter than the pore signal), (2) smoothness of trajectory, and (3) position relative to the nuclear centre. This latter criterion is necessary because Nup49-GFP staining can be confused with a weak perinuclear locus. All three parameters can be modulated individually in order to optimise the tracking for different situations (loci that are more mobile, more peripheral, of variable intensity, etc). Most importantly, the program has the option of further constraining the optimisation by forcing the trajectory to pass through a manually defined pixel. In that way mistracked spots can again be added manually to the correct trajectory, which is recalculated quasi-instantaneously. This tracking method proves to be extremely robust and reproducible due to its global approach.
Note: Some commercially available software are also able to track objects [e.g., Imaris (Bitplane), Volocity (Improvision)], although tracking efficiency is variable and usually requires uniformly highquality images. The algorithms are mostly based on threshold principles, which are rarely modifiable or interactive, and which are ill-suited for noisy images.
Because each time-lapse series represents a single cell, it is indispensable to average 8-10 movies over a total time >40min for a given strain or condition. Subtle differences require a larger data source. Useful parameters for quantitative analysis include the following.
- Track length. The projected track of the tagged locus can be visualised using LSM software, ImageJ, Excel, or other programs (Fig. 5A). The sum of all 1.5- s step lengths within a time-lapse series yields the total track length of that movie. From this, average track length and velocity (µm/min) can be calculated, but often this parameter is not very revealing.
- Step size. A histogram of step size distribution describes the nature of the movement more precisely. Statistical parameters such as mean, median, and standard deviation of individual and groups of movies can be calculated and compared with statistical tests (e.g., ANOVA). Even small but reproducible significant differences can be documented due to the large number of measurements.
- Large movements. Often differences in mobility are not obvious by comparing average speed, yet the frequency of large steps >500nm will vary significantly. These indicate transient high velocity movements. We generally score for steps larger than 500nm during seven frames (10.5 s), an interval that has proven useful for distinguishing patterns of mobility between different physiological states and stages of the cell cycle (Heun et al., 2001b). These are reported as the number of large steps per 10 min, averaged over at least 50 min of time-lapse imaging. Although a 500nm is a meaningful cutoff, any threshold over 300nm can be used.
- Mean square displacement (MSD). Observing the movement of a DNA locus over time not only gives information about its velocity, but also about the subvolume of the nucleus that it occupies during a given period of time. It has been shown for several chromosomal loci that chromosomal domains are able to move apparently randomly in a given subvolume (Gasser, 2002). This constraint can be quantified by MSD analysis, assuming that the movement of the spot follows a random walk. Ideally it describes a linear relationship between different time intervals and the square of the distance travelled by a particle during this period of time (MSD or (Δd2), where Δd2 = {d(t) - d(t + Δt)}2 (Berg, 1993; Marshall et al., 1997; Vazquez et al., 2001). In order to get the numbers, one must calculate the distances travelled by the spot for each time interval (1.5, 3, 4.5s...) and plot the square of the mean against increasing time intervals. These calculations and the corresponding graphs can be performed easily in Excel (Microsoft) or Mathematica (Wolfram Research). A representative MSD graph is shown in Fig. 5B. In these curves, the slope reflects the diffusion coefficient of the particle, and the linearity of the curve is usually lost at larger time intervals due to spatial constraint on the freedom of movement of the locus, i.e., the random walk of the particle is obstructed by the nuclear envelope or other subnuclear constraints, leading to a plateau (horizontal dashed line in Fig. 5B). The height of this plateau is related to the volume in which the particle is restricted. The slope of the MSD relation is directly correlated with diffusion coefficient. As explained earlier, in enclosed systems, the diffusion coefficient decreases with increasing At due to space constraints exerted on the particle dynamics. Nevertheless, the maximal diffusion coefficient can be calculated for very short time intervals and reflects the intrinsic mobility of particles (see sloping dashed line in Fig. 5B). For chromosomal loci in yeast, we observed a maximal diffusion coefficient in the range of 1 × 10-4 to 1 × 10-3µm2/s based on short time intervals. If distances are measured between two separate moving loci, <Δd2> reflects two times the MSD of an individual spot or locus moving relative to a fixed point (Vazquez et al., 2001). A more theoretical discussion of these parameters is found in Berg (1993).
![FIGURE 5 Analysis of DNA locus dynamics. (A) The projected trace of 300 images of a movie of the LYS2 locus. The average track length in 5min is 37.4µm. Bar: 1 µm. (B) A mean square displacement (MSD, <Δd2> in [µm2]) analysis on an average of 8 movies of the LYS2 locus. All cells were observed in G1 phase.](images/v2_pb_s09_c46_f05.jpg) |
| FIGURE 5 Analysis of DNA locus dynamics. (A) The projected trace of 300 images of a movie of the LYS2 locus. The average track length in 5min is 37.4µm. Bar: 1 µm. (B) A mean square displacement (MSD, <Δd2> in [µm2]) analysis on an average of 8 movies of the LYS2 locus. All cells were observed in G1 phase. |
It is very difficult to accurately quantify the intensity of a small, mobile GFP-laci focus. Even in deconvolved images it can differ by twofold in sequential images.
This protocol shows the optimal method for the described microscope setups. For different microscopes, the values and methods of this protocol are simply a starting point for further optimisation. As improvements in technology (e.g., more sensitive and rapid CCD cameras) and reagents (e.g., more stable or more intense GFP variants) evolve, future adjustments of this protocol will be indispensable.
The method described here can also be applied to Schizosaccharomyces pombe with a few changes, one being immobilisation on a coverslip with isolectin B (1 mg/ml) (Williams et al., 2002) or lectin from Bandeiraea simplicifolia (lyophilized powder, Sigma Cat. No. L2380).
V. PITFALLS
- To ensure that DNA movements are not the result of nuclear rotation, fluorescence recovery after photobleaching on GFP nuclear pore components should be performed over the same time intervals used to monitor DNA movement.
- To increase oxygen concentration and to prevent massive production of CO2 under the cover slide, vortex the agarose/medium before making the patch.
- Growth conditions must be standardised thoroughly, because both choice of carbon source and its concentration significantly influence subnuclear position and dynamics of tagged loci.
- Cells grown in minimal medium may not pellet as well as cells grown in YPD. Concentrate cells by centrifuging 2 volumes of culture in the same 1.5-ml tube.
- In the Ludin chamber yeast cells often bud upwards into the medium (i.e., parallel to the optical axis). Thus it is important to scan the entire cell in transmission mode not to miss the presence of a bud.
- Observations made on individual cells are often not representative of entire populations. It is crucial to verify observed differences with the appropriate statistical tests.
Belgareh, N., and Doye, V. (1997). Dynamics of nuclear pore distribution in nucleoporin mutant yeast cells. J. Cell Biol. 136(4), 747-759.
Belmont, A. S. (2001). Visualizing chromosome dynamics with GFP. Trends Cell Biol. 11(6), 250-257.
Berg, H. C. (1993). "Random Walks in Biology." Princeton Univ. Press, Princeton, NJ.
Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2002). A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 99(12), 7877-7882.
Fuchs, J., Lorenz, A., and Loidl, J. (2002). Chromosome associations in budding yeast caused by integrated tandemly repeated transgenes. J. Cell Sci. 115(Pt 6), 1213-1220.
Gasser, S. M. (2002). Visualizing chromatin dynamics in interphase nuclei. Science 296(5572), 1412-1416.
Guthrie, C., and Fink, G. R. (1991). "Guide to Yeast Genetics and Molecular Biology." Academic Press, San Diego.
Hediger, F., Neumann, F. R., Van Houwe, G., Dubrana, K., and Gasser, S. M. (2002). Live Imaging of Telomeres: yKu and Sir Proteins Define Redundant Telomere-Anchoring Pathways in Yeast. Curr. Biol. 12(24), 2076-2089.
Heun, P., Laroche, T., Shimada, K., Furrer, P., and Gasser, S. M. (2001b). Chromosome dynamics in the yeast interphase nucleus. Science 294(5549), 2181-2186.
Lisby, M., Mortensen, U. H., and Rothstein, R. (2003). Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nature Cell Biol. 5(6), 572-577.
Marshall, W. F., Straight, A., Marko, J. F., Swedlow, J., Dernburg, A., Belmont, A., Murray, A. W., Agard, D. A., and Sedat, J. W. (1997). Interphase chromosomes undergo constrained diffusional motion in living cells. Curr. Biol. 7(12), 930-939.
Michaelis, C., Ciosk, R., and Nasmyth, K. (1997). Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 91(1), 35-45.
Rasband, W. ImageJ. National Institute of Health, Bethesda, MD.
Sage, D., Neumann, F. R., Hediger, F., Gasser, S. M., and Unser, M. (2005). "Automatic Tracking of Individual Fluorescent Particles: Application to Chromatin Dynamics." in IEEE Transactions on Image Processing., in press.
Straight, A. F., Sedat, J. W., and Murray; A. W. (1998). Time-lapse microscopy reveals unique roles for kinesins during anaphase in budding yeast. J. Cell Biol. 143(3), 687-694.
Vazquez, J., Belmont, A. S., and Sedat, J. W. (2001). Multiple regimes of constrained chromosome motion are regulated in the interphase Drosophila nucleus. Curr. Biol. 11(16), 1227-1239.
Williams, D. R., and McIntosh, J. R. (2002). mcl1+, the Schizosaccharomyces pombe homologue of CTF4, is important for chromosome replication, cohesion, and segregation. Eukaryot Cell 1(5), 758-773.
Support our developers




