Immunocytochemistry of Frozen and of Paraffin Tissue Sections
Immunocytochemistry of tissue sections can yield valuable information as to the location of antigens. Thus it can determine whether the antigen is ubiquitous or is present only in certain tissues. It can further determine whether all cells in a given tissue are positive for a given antigen or whether the antigen is restricted to one or a few specialized cell types within the tissue. Its uses are not limited to normal tissues, and testing of tumor tissues in immunocytochemistry can yield information important for determination of tumor type. As stated in the articles on immunocytochemistry of cultured cells, choice of antibodies and of fixation method can be of critical importance. New antibodies should be tested on both frozen sections and paraffin sections of a variety of tissues in which the antigen is thought to be present. In general, more antibodies will react on frozen sections than on paraffin sections; however, morphology is better preserved in the paraffin sections.
II. MATERIALS AND INSTRUMENTATION
A. Antibodies
See the article by Osborn in this volume. Establish working dilutions by running a dilution series. Usually monoclonal antibodies can be diluted 1:5 to 1:30, ascites fluid 1 : 100 to 1 : 1000, and polyclonal antibodies 1:20 to 1:40, but the dilutions depend critically on the detection technique that is employed. Dilute both primary and secondary antibodies as far as possible to save money and to avoid unspecific reactions.
B. Cryostat Use a Leica CM1900 rapid sectioning cryostat (www.leica-microsystems.com). A useful accessory is a freezing head with variable temperature control. Use a C-knife and resharpen when necessary.
C. Tissue-tek
OCT 4583 compound from Miles Laboratories.
Automatic machines are useful only for laboratories that process a large number of samples. Check equipment for paraffin embedding in the local pathology department before purchasing.
E. Sliding Microtomes
These are relatively inexpensive and can be used with disposable knives.
F. Water Baths for Histology
These are thermostatically controlled and relatively shallow, e.g., 20cm in diameter and 4cm deep.
G. Microscopes
A microscope equipped with the appropriate filters to view immunofluorescence specimens is important. To view peroxidase- or streptavidin-biotin-stained specimens only a simple light microscope is required.
III. PROCEDURES
A. Cryostat Sections
1. Freezing of Tissue Blocks
- Fill the inner beaker of the freezing apparatus with isopentane and the outer beaker with liquid nitrogen about 30min before freezing tissues (Fig. 1). Use reagent-grade isopentane. Measure the temperature and wait until isopentane reaches -120 to -130°C. At -155°C the isopentane will freeze.
- Dissect tissues. Cut into small blocks (~4-7mm) using a scalpel. Place block on a prenumbered square of paper with the surface that will be sectioned furthest away from the paper. For small specimens, e.g., vessels, place a drop of Tissue-tek on the paper and then add the tissue.
- Drop tissue blocks into iospentane. Leave for at least 30s. Remove blocks with plastic tweezers and transfer directly to plastic vials (scintillation vials 6× 2.5 cm work well) or metal cans (~3 × 3 cm) with screw tops that have been precooled on dry ice. Close vials and store in a -70°C freezer. Tissue blocks are stable for several years.
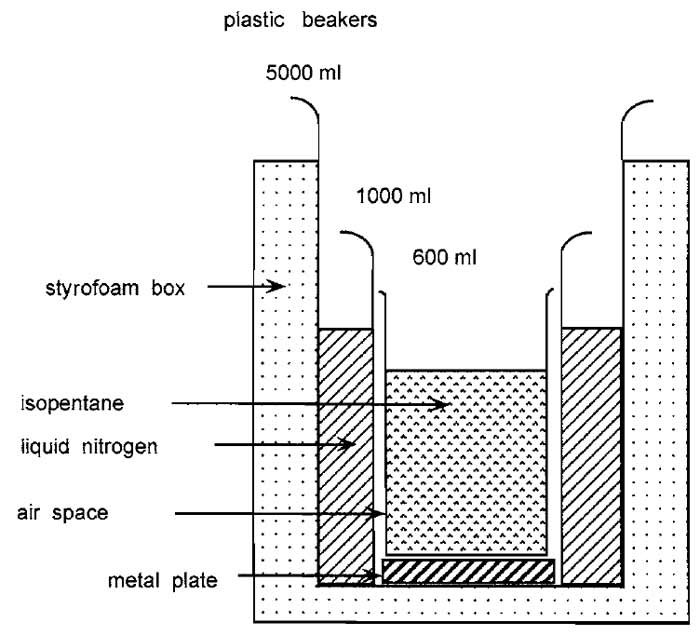 |
| FIGURE 1 Apparatus used to freeze tissues. |
2. Cutting Cryostat Sections
- Wash microscope slides by dipping in acetone, air dry, and store at room temperature. Store plastic tweezers and brush in the cryostat. Precool cryostat and freezing head.
- Place Tissue-tek on precooled freezing head and mount block so that the larger side of the cut section is at 90° to the knife blade.
- The optimal cutting temperature is different for different tissues. Most tissues cut well at -15 to -20°C. For liver, use -10°C. If the sections wrinkle as they are cut and look mushy, decrease the temperature. If the sections have cracks and look brittle, increase the temperature.
- To cut sections use the C-knife. First trim the block to get a good cutting surface. Then adjust the section thickness to 5µm and use the automatic advance. Now cut three or four sections so that the preset section thickness is achieved. Then bring the antiroll plate on the knife to stop the section from rolling up and to keep it fiat on the knife. The antiroll plate must be parallel to the knife edge and should only protrude very slightly over the edge.
- For optimal sections the knife and the antiroll plate must be kept clean (use a soft cloth dipped in acetone). Always clean the knife in the cutting direction and never the reverse so as not to damage the cutting edge of the knife.
- Remove the antiroll plate and hold the microscope slide over but not touching the cut section. The section should now spring on to the slide because of the difference in temperature between knife and slide. The quality of the section can be checked using toluidine blue or hematoxylin-eosin staining (see later).
- Dry the sections at room temperature for 30min. Then either use directly or place in a slide box and put in a -70°C freezer. Cut sections are stable for months or years at -70°C.
- Sections from a few tissues may not stick firmly enough to slides and may come off during subsequent processing. If this happens, try coating slides with 0.1% polylysine (e.g., Sigma Cat. No. 8920) in water.
B. Paraffin Sections
1. Embedding Tissues in Paraffin
Steps
Human material is often received from the clinic already embedded in paraffin. Protocols vary depending on the clinic, with time of fixation in formaldehyde being very variable (e.g., 4 h to over the weekend). To embed animal tissue in the laboratory, cut into 4 to 7- mm blocks and place it for 4-8 h in 3.7% formaldehyde in phosphate-buffered saline (PBS), 1 h in 50% ethanol, 2 × 1 h in 70% ethanol, 2 × 1 h in 96% ethanol, 2 × 1 h in 100% ethanol, 1 × 1 h in xylene, 1 × 2 h in xylene, and 2 × 2 h in Paraplast Plus (Shandon).
2. Cutting Paraffin Sections
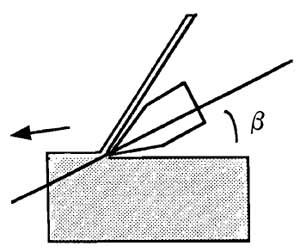 |
| FIGURE 2 Correct adjustment of knife and of cutting angle to cut paraffin sections. |
Steps
- To obtain very thin sections (1-2µm), put the paraffin blocks in a freezer at -20°C for about 30min. Mount the block in the holder of a sliding microtome. If the block cuts well do not use the automatic advance, but rely instead on the natural expansion of the block as it warms up to advance the block. If the block is not easy to cut, use the automatic advance set at a thickness of 1-2 µm. Correct adjustment of the knife and of the cutting angle is very important. Use an inclination angle (β) of 15° (Fig. 2). If the inclination angle is less than 10° the knife will not cut the block, and if it is greater than 15° the block will break.
- Trim the block until the cutting surface is optimal. Then cut a section, using a paint brush to draw the section onto the knife so that it does not roll up.
- Dip a second paint brush in water so that the section will adhere to it and move the section to a water bath held at 40-45°C. If the section is placed with the shiny smooth surface touching the water the warmth will smooth out the section and wrinkles will vanish!
- Place a microscope slide under the section and, using a brush, position the section on the slide.
- Dry the sections. For immunohistochemical methods, dry overnight at 37°C. For normal histological methods, set drying oven to 60°C so that the paraffin melts in part during the drying step and dry for 1-2 h.
3. Deparaffinization
Immerse sections 2 × 10min in xylene, 1 × 3 min in 100% ethanol, 1 × 3 min in 95% ethanol, and air dry.
Some laboratories routinely use trypsinization or other proteolytic treatment of formalin-fixed, paraffinembedded tissues prior to immunohistochemistry, e.g., 5min in 0.1% trypsin (Sigma Cat. No. T-8128 or Dako Cat. No. $2012) in PBS at room temperature. As stressed by Ordonez et al. (1988), this can enhance the staining by certain antibodies but may also result in false negative staining with other antibodies. Thus, control trypsinization conditions carefully and recheck them each time a new antibody is used.
5. Treating Sections in a Microwave Oven
Cut sections onto Superfrost or similar slides. After deparaffinization, immerse slides in citrate buffer (2.1 g citric acid monohydrate/liter adjusted to pH 6.0 with NaOH). Microwave for two cycles of 5 min each at a setting of 650 or 700 W. Add more buffer between cycles so slides stay covered during the microwave step. Cool to room temperature (cf. Cattoretti et al., 1992).
C. Histologic Staining of Sections
Stain frozen sections directly. Deparaffinize paraffin sections (see earlier), substituting a wash with distilled water for the air-drying step.
- Toluidine blue: Immerse sections in 1% toluidine blue for approximately l min. Wash with distilled water. Mount in water-soluble embedding medium, e.g., Glycergel (Dako Cat. No. C0563).
- Hemotoxylin-eosin: Immerse sections for 10 min in Mayer's Hemalum solution (Merck). Wash for 10 min under running tap water, for 5 min in eosin (Merck), and twice with distilled water and then run through an alcohol series (e.g., 75%, 95% for 2 min each, then 100% for 5 min, then 2 × 5 min in xylol) and mount in Eukitt (Riedel de Ha6n Cat. No. 33949) or Entellan (Merck Cat. No. 107960).
D. Immunocytochemistry
Three methods are described in detail: immunofluorescence, the immunoperoxidase method, and the more sensitive streptavidin-biotin method. The two latter methods have the advantage that nuclei can be counterstained with hemotoxylin and that only a simple light microscope is required to visualize the stain; however, fluorescence generally gives greater resolution.
As noted for cells, many antibodies that react well on cryostat sections may not react on the same tissue after it has been fixed in formaldehyde and embedded in paraffin. In such a case it may be advantageous to try fixing tissue, e.g., in B5, Bouin's, or Zenker's fixative or alcohol, prior to paraffin embedding. An interesting alternative is to use sections of formaldehyde-fixed, paraffin-embedded material that have been treated in a microwave oven.
For all methods mark the position of the section after fixation; either use a diamond pencil or circle the section with a water-repellent marker (Dako Cat. No. S2002). Remove excess buffer after rinsing steps with Q-tips. Use 10µl of antibody per section. Apply with an Eppendorf pipette and use the pipette tip to spread the antibody over the section without touching the section. Several manufacturers (e.g., Dako) produce excellent protocol sheets for each immunocytochemical method.
1. Immunofluorescence
Steps
- Fix cryostat sections or paraffin sections deparaffinized as described earlier for 10min in acetone at -10°C. Air dry.
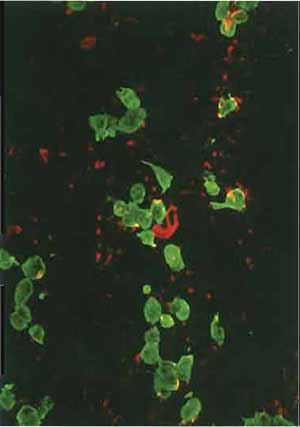
- Use steps 2-9 of the protocol given in the article by Osborn in this volume for multitest slides. Nuclei can be counterstained with Hoechst dye (see Osborn's article). Positively stained cells will be green (see Fig. 3) if an FITC-labeled second antibody is used or red if a rhodamine-labeled second antibody is selected (see also the double label of a cytological sample in Fig. 4).
|
|

Steps
- Fix cryostat sections for 10 min in acetone at -10°C, air dry, and wash in PBS.
- Deparaffinize paraffin sections and incubate for 30 min at room temperature in 100ml methanol containing 100µl H2O2 to block endogenous peroxidase activity. Wash in PBS.
- Incubate for 10min at 37°C with normal rabbit serum. Drain, but do not wash after this step.
- Incubate with primary antibody (e.g., mouse monoclonal) for 30min at 37°C.
- Wash three times in PBS.
- Incubate with second antibody coupled to peroxidase, e.g., rabbit antimouse for a monoclonal first antibody (Cat. No. P0260 from Dako diluted 1:10 to 1:20).
- Wash three times in PBS and once in Tris buffer (6 g NaCI, 6 g Tris/liter, pH 7.4),
- Develop for 10min at room temperature using freshly made solutions (e.g., 0.06g diaminobenzidine, Fluka Cat. No. 32750 in 100ml Tris buffer, 0.03ml H2O2). Note: Diaminobenzidine is a carcinogen; handle with care.
- Wash in tap water.
- Apply a light counterstain by immersing the slide in Hemalum for 1 to 10s. Remove when staining reaches the required intensity.
- Wash in tap water.
- Mount in Glycergel (Dako Cat. No. C0563, http://www.dako.com).
Notes
- Structures that are positively stained will be dark brown, whereas nuclei will be light blue (Figs. 5-7).
- The method can be made more sensitive by using an additional step with a peroxidase-antiperoxidase complex (see Sternberger, 1979).
- Blocking should be performed with a 5% solution of normal serum from the same host species as the labeled antibody, i.e., in this example the tissue is blocked with normal rabbit serum because the labeled antibody is a rabbit antibody.
- Bovine serum albumin (BSA) for diluting antibodies should be of high purity. Impure preparations can contain bovine IgGs that can cross-react with the labeled second antibodies.
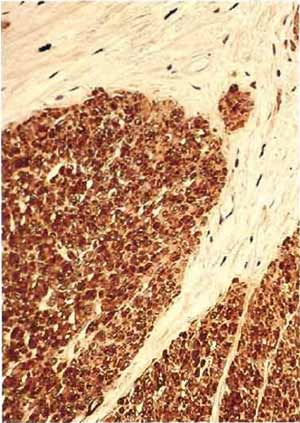 |
| FIGURE 5 Paraffin section of human uterus stained with antibody after microwave fixation with the desmin DER 11 antibody in the peroxidase technique (×160). |
|
|
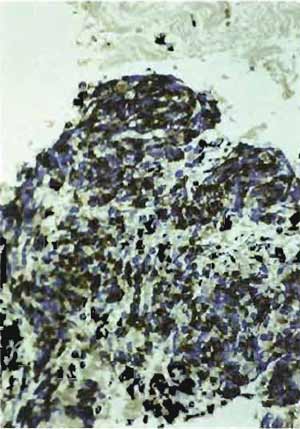
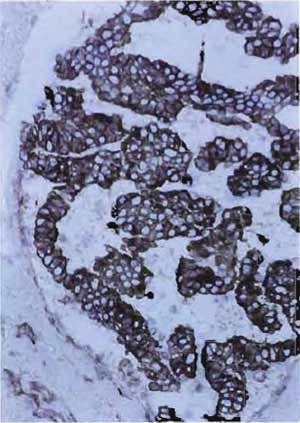
3. Streptavidin-Biotin Stain
Buy the reagents separately or use the Histostain kit from Zymed Laboratories (http://www.zymed.com; Cat. No. 95-6543 for mouse primary antibody and Cat. No. 95-6143 for rabbit primary antibody). These kits are based on the strong binding between streptavidin, a 60,000-kDa protein isolated from Streptomyces avidinii, and biotin, a water-soluble vitamin (MW 244, Kd = 10-15M). Instructions are given for the mouse kit.
- Fix cryostat sections for 10 min in acetone at -10°C and air dry. Sections can be treated with 0.23% periodate for 45s. Go to step 3.
- Deparaffinize paraffin sections and air dry. Incubate 10min in PBS at room temperature and then 10min in H2O2 solution (nine parts methanol to one part 30% H2O2 in water). Wash 3 x 2min in PBS.
- Reduce nonspecific background staining by blocking for 10min in 10% goat serum at room temperature (see Note 3 in Section III,D,2). Then drain but do not wash.
- Incubate with primary antibody, e.g., mouse monoclonal antibody, in wet chamber for 30min at 37°C.
- Wash 3 × 2 min in PBS.
- Add biotinylated second antibody, e.g., goat antimouse, for 10min at room temperature and repeat step 5.
- Add enzyme conjugate streptavidin-peroxidase diluted 1:20 for 5min at room temperature. This binds to the biotin residues on the second antibody.
- Develop by adding substrate-chromogen mixture for 5min at 37°C or 15min at room temperature. The enzyme peroxidase catalyzes the substrate hydrogen peroxide and converts the chromogen aminoethylcarbazole to a red, colored deposit.
- Wash 3 × 2 min in distilled water.
- Counterstain with Hemalum between 1 and 10 s.
- Wash 7min in tap water.
- Mount in glycergel.
Notes
- Structures that are positively stained will be red, whereas nuclei will be light blue (Figs. 8 and 9).
- The optimal dilution of primary antibody has to be determined for each antibody.
- The streptavidin-peroxidase conjugate is the same for all species; the blocking serum and second antibody vary according to the species in which the first antibody is made.
|
|
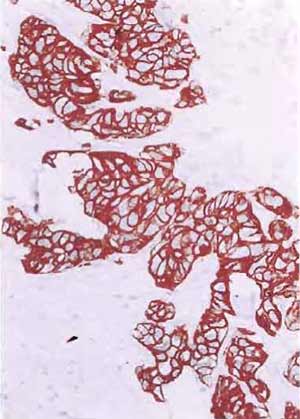
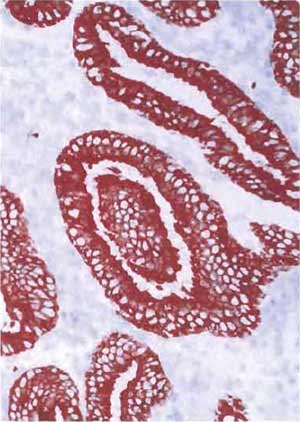
Other Methods
- The alkaline phosphatase-antialkaline phosphatase technique is also a very sensitive method (Fig. 8).
- Both fluorescent and chromogenic signals can be enhanced up to 1000X with Tyramide Signal Amplification Technology (New England Nuclear). With this technique, primarily antibodies can be diluted in some instances more than 10,000-fold.
If possible, include (1) a positive control, i.e., a section of a tissue known to contain the antigen; (2) a negative control, i.e., a section known not to have the antigen; and (3) a reagent control, i.e., a section stained with nonimmune serum instead of the primary antibody. Ideally, the first control should be positive and the second and third controls negative. If high backgrounds are obtained, it may help to adjust the antibody concentrations by increasing the length of time for washing steps or by including 0.5M NaCl or 5% BSA in the antibody solutions.
References
Cattoretti, G., Backer, M. H. G., Key, G., Duchrow, M., Schliiter, C., Galle, J., and Gerdes, J. (1992). Monoclonal antibodies against recombinant parts of the Ki-67 antigen (MIB 1 and MIB 3) detect proliferating cells in microwave processed formalin-fixed paraffin sections. J. Pathol. 168, 357-363.
Denk, H. (1987). Immunohistochemical methods for the demonstration of tumor markers. In "Morphological Tumor Markers" (G. Seifert, ed.), pp. 47-70. Springer-Verlag, Berlin.
Ordonez, N. G., Manning, J. T., and Brooks, T. E. (1988). Effect of trypsinization on the immunostaining of formalin-fixed, paraffin- embedded tissues. Am. J. Surg. Pathol. 12, 121-129.
Osborn, M., and Domagala, W. (1997). Immunocytochemistry. In "Comprehensive Cytopathology" (M. Bibbo, ed.), 2nd Ed., pp. 1033-1074. Saunders, Philadelphia.
Osborn, M., and Weber, K. (1983). Tumor diagnosis by intermediate filament typing: A novel tool for surgical pathology. Lab. Invest. 48, 372-394.
Sternberger, L. A. (1979). "Immunohistochemistry," 2nd Ed. Wiley, New York.
Tubbs, R. R., Gephardt, G. N., and Petras, R. E. (1986). "Atlas of Immunohistology." American Society of Clinical Pathologists Press, Chicago.
Support our developers




