Large Scale Protein Localization in Yeast
Proteomics has contributed significantly to our knowledge of eukaryotic cell biology through largescale studies describing protein-protein interactions, protein complex composition, and relative protein abundance. Recent work in the budding yeast Saccharomyces cerevisiae (Kumar et al., 2002) has extended the scope of proteomics to encompass the genome-wide analysis of protein localizationmthe large-scale and systematic identification of the subcellular compartment or organelle to which a protein localizes. The subcellular localization of a protein can be of fundamental importance in characterizing the function and regulation of that protein. For example, an uncharacterized protein that localizes to the mitochondria may be inferred to function directly or indirectly in cellular respiration. Many transcription factors are regulated through subcellular compartmentalization: nuclear export of the transcription factor Pho4p is tightly regulated such that it is only localized within the nucleus (and therefore capable of activating transcription) in response to phosphate starvation (O'Neill et al., 1996). Protein function can also be addressed on a global scale by integrating protein localization data with a variety of other proteomic data sets. In particular, protein localization data are a strong complement to large-scale protein-protein interaction studies, corroborating many putative interactions while also highlighting potential false-positive results (Gerstein et al., 2002). Ultimately, genome-wide protein localization data will be helpful in identifying the constituent proteins of each cellular organelle and, potentially, mechanisms by which some of these proteins are regulated.
This article presents detailed protocols by which yeast ORFs may be epitope tagged and yeast strains containing tagged proteins prepared for immunofluorescence analysis in a 96-well format. The protocols presented here utilize the PCR-based tagging method of Longtine et al. (1997) to generate C-terminal chromosomal gene fusions with a sequence encoding three copies of the hemagglutinin (HA) epitope. The resulting HA-tagged yeast proteins can be immunolocalized by indirect immunofluorescence as described; the immunofluorescence protocol is adapted for performance in a 96-well format, facilitating proteome-scale analysis of protein localization.
G418 sulfate (geneticin) may be purchased from Invitrogen Corp. (Cat. No. 11811023). Glusulase is a trademarked preparation of β-glucuronidase / sulfatase, each available from Sigma (Cat. Nos. G0258 and S8504). Zymolyase-100T is from Seikagaku America; Lyticase from Sigma may be substituted (Cat. No. L4025). The mouse anti-HA monoclonal antibody 16B12 is from Covance (Cat. No. MMS-101R-1000). Cy3-conjugated affinity-purified goat antimouse IgG is from the Jackson Laboratories (Cat. No. 115-165-146). 4,6-Diamidino-2-phenylindole (DAPI) is available from Sigma (Catalog No. D-9542). Poly-L-lysine is from Sigma (Cat. No. P1524). Microscope slides are from Carlson Scientific (Cat. No. 101805).
III. PROCEDURES
A. Epitope-Tagging Yeast Genes
Yeast genes may be epitope tagged through a variety of methods: the protocol described here is essentially that of Longtine et al. (1997) in which PCR is used to amplify a cassette from the vector pFA6a- 3HA-kanMX6 suitable for generating an integrated Cterminal HA-tagged allele of any nonrepeated yeast gene. The HA-tagging cassette (Fig. 1A) consists of a sequence encoding three copies of the HA epitope, the S. cerevisiae ADH1 terminator, and the kanMX6 module encoding resistance to G418. Tagging cassettes are amplified using primers with 5' ends corresponding to the target gene and 3' ends corresponding to the cassette; primer design is discussed later. Amplified cassettes are subsequently introduced into yeast by standard methods of DNA transformation (Gietz et al., 1992). By homologous recombination, the tagging cassette integrates at its intended genomic locus; transformants containing this cassette are selected on medium supplemented with G418. By this PCR-based approach, yeast genes may be systematically epitope tagged for subsequent large-scale immunolocalization.
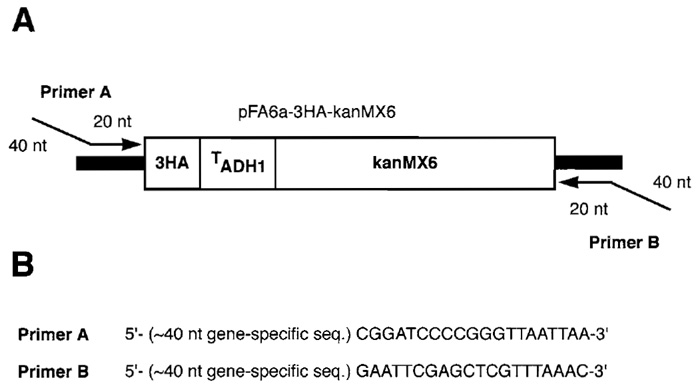 |
| Figure 1 (A) HA-tagging cassette amplified from the yeast vector pFA6a-3HA-kanMX6. The tagging cassette contains a sequence encoding three copies of the influenza virus hemagglutinin epitope (3HA), the S. cerevisiae ADH1 terminator, and the sequence encoding the kanMX6 cassette (encoding resistance to G418 sulfate). PCR primers for amplification of this cassette are designed such that the 5' end of each primer consists of approximately 40 nucleotides of a gene-specific sequence, whereas the 3' end consists of approximately 20 nucleotides of sequence from the polylinker immediately flanking the tagging cassette. (B) Example of PCR primers suitable for C-terminal HA tagging of target genes. |
- YPD medium: 1% Bacto-yeast extract, 2% Bactopeptone, 2% glucose, 2% Bacto-agar, distilled water. Autoclave at 121°C and 15lb/inz. in of pressure for 15 min; cool to ~55°C and pour plates. Omit agar for liquid media.
- YPD-G418 plates: Prepare YPD medium. After cooling, supplement YPD with 150mg/liter G418. To prepare a stock solution of G418 (geneticin), dissolve in water at a concentration of 80mg dry weight per milliliter and filter sterilize.
Steps
- Design primers enabling targeted integration of the amplified tagging cassette at its intended genomic locus. Example primers are shown in Fig. 1B. Design the 5' end of each primer such that it consists of 40-50 nucleotides of sequence from the target gene. Choose the gene-specific sequence of the forward primer (A) such that it ends just upstream of the stop codon; choose the gene-specific sequence of the reverse primer (B) such that it ends just downstream of the stop codon.
- Amplify the HA-tagging cassette from plasmid pFA6a-3HA-kanMX6 using PCR primers A and B. Use standard cycling conditions; vary conditions and primer/template concentrations as needed. Repeat PCR reaction four or more times. Pool reactions, extract once with phenol: chloroform:isoamyl alcohol (25:24:1), precipitate, and resuspend in a small volume of water (~10µl).
- Introduce concentrated PCR product into S. cerevisiae by any standard DNA transformation protocol (e.g., the lithium acetate protocol of Gietz et al., 1992). This process may be performed in 96-well format as described previously (Kumar et al., 2000).
- Select transformants on YPD medium supplemented with G418. Wash cells in water (~1ml), and resuspend in 200 µl of water. Spread onto YPD plates; incubate overnight at 30°C and replicate onto YPDG418 plates. Pick individual G418-resistant colonies. Alternatively, G418 selection may be performed in large scale. Wash, precipitate, and resuspend transformants in 400 µl YPD in 96-well plates. Incubate at 30°C for 4-6h with shaking. Streak transformant mixtures onto rectangular 86 x 128-mm YPD plates supplemented with G418. Use an eight-pronged replicator, such that 24 streaks (correponding to 24 transformant cultures) may be produced per plate. Pick G418- resistant colonies as described previously.
- Using PCR, confirm that the HA-tagging cassette has integrated by homologous recombination with the target gene. Choose one primer such that it anneals within the HA-tagging cassette and another primer that anneals to the target gene locus outside of the insertion region. Identify a PCR product of the expected size as evidence of homologous integration.
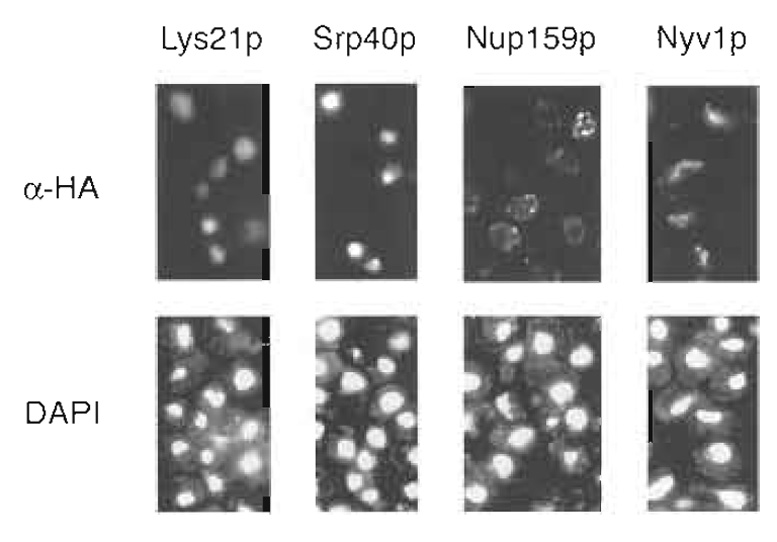 |
| Figure 2 Examples of immunofluorescence patterns from largescale protein localization studies in S. cerevisiae. (Top) Immunofluorescence patterns in vegetative yeast cells stained with monoclonal antibodies directed against HA. (Bottom) The same cells stained with the DNA-binding dye 4, 6-diamidino-2-phenylindole (DAPI). Staining of the nucleus is evident in cells carrying a tagged allele of the homocitrate synthase Lys21p, while nucleolar staining can be seen in cells HA tagged for the chaperone Srp40p. HA-tagged Nup159p localizes to the nuclear rim, while staining of the vacuole may be observed in cells containing a tagged form of the v-SNARE Nyvlp. |
Strains containing HA-tagged proteins may be immunolocalized by indirect immunofluorescence using monoclonal antibodies directed against the HA epitope (primary antibody) and the Cy3-conjugated secondary antibody. Immunolocalization in yeast necessitates permeabilization of the cell wall; here, yeast cells are spheroplasted using the trademarked enzyme preparations glusulase and Zymolyase-100T. The provided immunolocalization protocol is tailored for implementation in a 96-well format. Using this procedure, a single researcher can easily prepare 192 samples (two 96-well microtiter plates) for immunofluorescence in a period of 3 days (Kumar et al., 2000). Sample immunofluorescence patterns in cells containing HA-tagged proteins are presented in Fig. 2.
- Solution A: 1.2M sorbitol, 50mM KPO4, pH 7. Prepare sorbitol as a 2M stock solution in water. Store solution A up to 1 week at 4°C.
- Solution A / glusulase / Zymolyase: Supplement solution A with 1 µl/ml 2-mercaptoethanol, 1 µl/ml glusulase, 1µl/ml Zymolyase (at a concentration of 10mg/ml). Prepare a stock solution of 10mg/ml Zymolyase in 5% glucose in 20mM phosphate buffer, pH 7.4. Store stock solution of Zymolyase at -20°C. Add 2-mercaptoethanol under a chemical fume hood. Prepare this mixture immediately before use.
- Phosphote-buffened Saline (PBS) buffer: 20x PBS buffer per liter, 160g NaCl, 4g KCl, 28.8g Na2HPO4, 4.8g KH2PO4, 800ml water. Adjust pH to 7.4; adjust volume to 1 liter. Autoclave and store at room temperature. Dilute 20-fold prior to use. PBS buffers supplemented with BSA may be stored for approximately 1 week at 4°C.
- Antibody solutions: Primary and secondary antibodies should be added to PBS buffer immediately prior to use. Centrifuge antibodies at 4°C for 10min at maximum speed in a variable-speed microcentrifuge (e.g., Eppendorf Model 5415C centrifuge). Store the Cy3-conjugated secondary antibody in the dark at -20°C.
- Poly-L-lysine solution: 0.5mg/ml poly-L-lysine. Dissolve 50mg poly-L-lysine powder in 10ml H2O; dilute 10-fold in H2O. Filter sterilize if desired and aliquot into 1.5-ml tubes. Store at -20°C. Centrifuge poly-L-lysine solution for 10min prior to use.
- DAPI stock solution: Dissolve DAPI at a concentration of 1 mg/ml in sterile H2O. Wear gloves and safety glasses when working with DAPI, as it is a possible carcinogen. Store at -20°C.
- Mount medium with DAPI: 1mg/ml p-phenylenediamine in PBS supplemented with DAPI (final concentration of 1 µg/ml). Dissolve 100mg p-phenylenediamine in 10ml PBS buffer; prepare solution in a small beaker covered with aluminum foil. Add 90ml glycerol and continue stirring. Aliquot into 1.5-ml tubes and store at -70°C. Before use, add 1µl DAPI stock / ml mount medium. Mount medium with DAPI may be stored for up to 2 weeks.
- Inoculate strains containing HA-tagged proteins in 96-well microtiter plates containing 75 µl YPD media per well. Grow cultures overnight (approximately 12 h) at 30°C with gentle shaking on an orbital platform shaker (240rpm). Alternatively, incubate plates on a vortex shaker set to its lowest speed. After overnight growth, add 45µl YPD; grow for an additional 90min to an OD600 of 0.75-1.
- Fix cells in formaldehyde at a final concentration of 3.75% (v/v). Agitate cells gently for 30min using a vortex shaker set to its lowest speed. Collect cells by centrifugation at 2000rpm for 4min in a Sorvall H-1000B rotor. Wash pelleted cells three times in 100µl solution A. Resuspend cells in 100 µl of solution A supplemented with 0.1% (v/v) 2-mercaptoethanol, 0.02% (w/v) glusulase, and Zymolyase-100T (5 µg/ml).
- Spheroplast cells in this mixture at 37°C with gentle shaking for 13-20 min (optimal incubation times must be determined empirically). Check cells periodically during this incubation; once translucent, recover cells and wash in 100 µl solution A. Resuspend pelleted cells in 100t, tl PBS buffer supplemented with 0.1% (v/v) Nonidet P-40 and 0.1% (w/v) bovine serum albumin (BSA). Incubate samples at room temperature without shaking for 15 min. Pellet cells by centrifugation as before. Remove excess liquid by aspiration, and resuspend pellets in 100 µl PBS supplemented with 3% (w/v) BSA. Gently shake cells at room temperature for 30 min to 1 h.
- Add 40µl of mouse anti-HA monoclonal antibody 16B12 at a final dilution of 1:1000 in PBS supplemented with 3% (w/v) BSA. Incubate overnight at 4°C with gentle shaking.
- Wash cells as follows: once in 150µl PBS supplemented with 0.1% (w/v) BSA, once with 100µl PBS supplemented with 0.1% (w/v) BSA and 0.1% (v/v) Nonidet P-40, and once in 100µl PBS supplemented with 0.1% (w/v) BSA. During this final wash, gently shake cells for 5min at room temperature prior to centrifugation.
- Treat pelleted cells with Cy3-conjugated affinitypurified goat antimouse IgG (secondary antibody) at a final dilution of 1:200 in 40µl of PBS supplemented with 3% (w/v) BSA. Shake cells gently at room temperature for 2 h in the dark (to minimize Cy3 exposure to light).
- Prepare poly-L-lysine-coated slides for use in step 9. To prepare two slides, add a total volume of 40µl poly-L-lysine solution (0.5mg/ml in H2O) to one slide; distribute the poly-L-lysine in five or six wells spaced evenly over the surface of the slide. Place the other slide (with no added polylysine) onto the poly- L-lysine-treated slide such that the two faces of the slides are sandwiched together. Wait 15 min. Separate slides and rinse in H2O; allow slides to air dry before use in step 9.
- Wash cells (from step 6) as follows: once in 150µl PBS plus 0.1% (w/v) BSA, once in 100 µl PBS plus 0.1% (w/v) BSA with gentle shaking for 5min, twice in 100µl PBS plus 0.1% (w/v) BSA and 0.1% (v/v) Nonidet P-40, and once in 100µl PBS plus 0.1% (w/v) BSA.
- Resuspend cells in 75 µl PBS. Using a multichannel pipetter, transfer 15µl of this cell suspension into 12 individual wells on a poly-L-lysine-treated slide. Allow cells to sit for a minimum of 15min before removing excess solution by aspiration. Add mounting medium supplemented with DAPI such that a total volume of 40µl is spotted at five or six locations in between the wells. Place a coverslip on the slide; avoid air bubbles and ensure that mounting medium is spread over all wells. Seal slides with nail polish and store in the dark at -20°C do not store slides longer than 1 month prior to examination.
In designing protein localization studies, consideration should be paid to the placement of reporter/epitope tags relative to each target open reading frame. Typically, reporters and epitope tags are fused to either N or C termini of target genes; however, each tagging strategy possesses associated advantages and drawbacks. Organelle-specific targeting signals (e.g., mitochondrial targeting peptides and nuclear localization signals) are often located at the N terminus (Silver, 1991). N-terminal reporter fusions may disrupt these sequences, resulting in aberrant protein localizations. In other cases, C-terminal sequences may be important for proper function and regulation. For example, isoprenylation motifs are typically C-terminal, and sequences important for plasma membrane localization may be found throughout the gene-coding sequence (Nakai and Horton, 1999). While no single tagging strategy can be all-encompassing, a greater proportion of the yeast proteome should remain functional as C-terminal fusions: several studies have generated genome-wide Cterminal fusions for large-scale protein localization (Ding et al., 2000; Kumar et al., 2002).
Gene copy number may also impact upon the accuracy with which a tagged protein is localized. Overexpressed protein products can saturate intracellular transport mechanisms, potentially resulting in anomalous protein localization. However, weakly expressed single copy genes may yield insufficient protein levels for analysis, although the protocol presented here-in which tagged proteins are expressed endogenously under control of their native promoters-has been used successfully to localize nonabundant proteins (Kumar et al., 2002).
References
Adams, A., Gottschling, D. E., Kaiser, C. A., and Stearns, T. (1998). Methods in Yeast Genetics, pp. 135-137. Cold Spring Harbor Laboratory Press, Plainview, NY.
Ding, D. Q., Tomita, Y., Yamamoto, A., Chikashige, Y., Haraguchi, T., and Hiraoka, Y. (2000). Large-scale screening of intracellular protein localization in living fission yeast cells by the use of a GFP-fusion genomic DNA library. Genes Cells 5, 169-190.
Gerstein, M., Lan, N., and Jansen, R. (2002). Proteomics: Integrating interactomes. Science 295, 284-287.
Gietz, D., St. Jean, A., Woods, R. A., and Schiestl, R. H. (1992). Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20, 1425.
Kumar, A., Agarwal, S., Heyman, J. A., Matson, S., Heidtman, M., Piccirillo, S., Umansky, L., Drawid, A., Jansen, R., Liu, Y, Cheung, K.-H., Miller, P., Gerstein, M., Roeder, G. S., and Snyder, M. (2002). Subcellular localization of the yeast proteome. Genes Dev. 16, 707-719.
Kumar, A., des Etages, S. A., Coelho, P. S. R., Roeder, G. S., and Snyder, M. (2000). High-throughput methods for the large-scale analysis of gene function by transposon tagging. Methods Enzymol. 328, 550-574.
Longtine, M. S., McKenzie, A., III, Demarini, D. J., Shah, N. G., Wach, A., Brachat, A., Philippsen, P., and Pringle, J. R. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953-961.
Nakai, K., and Horton, P. (1999). PSORT: A program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 24, 34-36.
Ross-MacDonald, P., Coelho, P. S. R., Roemer, T., Agarwal, S., Kumar, A., Jansen, R., Cheung, K.-H., Sheehan, A., Symoniatis, D., Umansky, L., Heidtman, M., Nelson, F. K., Iwasaki, H., Hager, K., Gerstein, M., Miller, P., Roeder, G. S., and Snyder, M. (1999). Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature 402, 413-418.
Ross-Macdonald, P., Sheehan, A., Roeder, G. S., and Snyder, M. (1997). A multipurpose transposon system for analyzing protein production, localization, and function in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 94, 190-195.
Silver, P. A. (1991). How proteins enter the nucleus. Cell 64, 489-497.
Support our developers




