Microsome-B, ased Assay for Analysis of Endoplasmic Reticulum to Golgi Transport in Mammalian Cells
The trafficking of proteins along the first stage of the secretory pathway is mediated by small vesicles that bud from the endoplasmic reticulum (ER) and subsequently fuse with the cis-Golgi compartment. This article describes a biochemical assay using mammalian microsomes that can be used to measure these events independently. The microsomes are prepared from cells infected at the restrictive temperature (39.5°C) with the ts045 strain of vesicular stomatitis virus (VSV) (Lafay, 1974). As a reporter molecule the assay utilizes ts045 VSV-glycoprotein (VSV-G), which is retained in the ER during infection due to a thermoreversible folding defect; incubation in vitro at the permissive temperature (32°C) results in the synchronous folding and transport of VSV-G to the Golgi complex. To follow vesicle formation, a differential centrifugation procedure is employed to separate the more rapidly sedimenting ER and Golgi membranes from the slowly sedimenting vesicles. Consumption is analyzed using a two-stage assay in which vesicles isolated by differential centrifugation during stage 1 are subsequently added to stage 2 (fusion) reactions containing acceptor Golgi membranes. Transport to the Golgi is measured by following the oligosaccharide processing of VSV-G from the high mannose ER form, which is sensitive to endoglycosidase H (endo H), to the cis/medial-Golgi form, which is endo H resistant (Schwaninger et al., 1992). The biochemical characteristics of the overall ER to Golgi transport reaction and the vesicle formation and consumption assays are described elsewhere (Aridor et al., 1995,1996b, 1998,1999a,b, 2000, 2001; Rowe et al., 1996a).
Culture medium (α-MEM; Cat. No. 11900-099) is from Life Technologies. The medium is supplemented with penicillin/streptomycin from a 100× stock solution (Cat. No P0781; Sigma). Fetal bovine serum (FBS; Cat. No FB-01) is from Omega Scientific. D-Sorbitol (Cat. No. S-1876), leupeptin (Cat. No L-2884), chymostatin (Cat. No C-7268), pepstatin (Cat. No P-4265), phenylmethylsulfonyl fluoride (PMSF; Cat. No P- 7626), actinomycin D (Cat. No A-1410), uridine 5'- diphospho-N-acetylglucosamine (UDP-GlcNAc; Cat. No. U-4375), and dimethyl sulfoxide (DMSO; Cat. No. D-2650), are from Sigma. The nitrocellulose membrane (Cat. No. 68260) is from Schleicher & Schuell. Horseradish peroxidase-conjugated goat anti-rabbit IgG (Cat. No. 31460) is from Pierce. Chemiluminescence reagent (Cat. No. NEL-101) and autoradiography film ("Reflection") are from NEN. Polyallomer microfuge tubes (Cat. No. 357448) are supplied by Beckman Instruments Inc. A polyclonal antibody to VSV-G is generated in rabbits immunized with the C-terminal 16 amino acids of VSV-G (Indiana serotype) coupled to KLH (Plutner et al., 1991). Centrifugation at 20,000 or 100,000g is performed using an Optima TL ultracentrifuge (Beckman) equipped with a TLA 100.3 rotor. A laser-scanning densitometer (personal densitometer; Molecular Dynamics) is used to quantitate data.
The following procedures are performed on ice unless otherwise stated.
A. Preparation of Cytosol
The following procedure for the preparation of rat liver cytosol is based on that described by Davidson et al. (1992).
Solutions
- Phosphate-buffered saline (PBS) (10× stock): 90mM phosphate and 1.5M NaCl (pH 7.4). To make 1 liter, add 80g NaCl, 2g KCl, 2g KH2PO4, and 21.6g Na2HPO4·7H2O to distilled water. Store at room temperature.
- 25/125: 0.125M KOAc, 25mM HEPES (pH 7.4). To make 100ml, add 3.125ml of 4M KOAc stock and 2.5 ml of 1M HEPES-KOH (pH 7.4) stock to distilled water. Store at 4°C.
- Protease inhibitor cocktail (PIC): 10µg/ml leupeptin, 10µg/ml chymostatin, 0.5µg/ml pepstatin A, and 1.0mM PMSE To supplement 50ml of buffer (e.g., 25/125) with PIC, add 50µl of 10mg/ml leupeptin stock (in H2O), 50 µl of 10 mg/ml chymostatin stock (in DMSO), 5µl of 5mg/ml pepstatin A stock (in DMSO), and 500µl of 0.1M PMSF stock (in ethanol). Use PIC buffer immediately or store at -20°C.
- Decapitate two anesthetized adult Sprague- Dawley rats (~250g), remove the livers, and place the tissue in a 250-ml glass beaker. Determine the weight of the tissue (typically ~20 g) and then wash two times with -50 ml of Ix PBS and once with ~50 ml of Ix PBS and once with ~50ml of 25/125.
- Finely mince the tissue using a pair of scissors and then homogenize in 2-3 volumes (ml/g of tissue) of 25/125 (PIC) with 20 strokes using a 40-ml Dounce (Wheaton). Use a "loose-fitting" Dounce for the first 10 strokes followed by a "tight-fitting" one for the second 10 strokes.
- Pour the homogenate into a 38-ml polycarbonate tube (Nalgene; Cat. No. 3117-0380) and centrifuge for 10min at 12,000g (10,000 rpm) in a Beckman JA20 rotor (Beckman Instruments Inc.). Using a pipette, transfer ~13 ml of the supernatant into each of two 14 x 89-mm ultraclear centrifuge tubes (Beckman Cat. No. 344059) and centrifuge at 150,000 g (35,000 rpm) for 90 min in a Beckman SW41 rotor.
- After centrifugation, remove the overlying lipid layer by aspiration and then withdraw the remaining supernatants (cytosol) from each tube using a pipette.
- Divide the cytosol into 250-µl aliquots in 0.5-ml microfuge tubes, freeze in liquid N2, and store at -80°C. The protein concentration of the cytosol is ~25 mg / ml.
Solutions
- Actinomycin D (200× stock): Add 10mg actinomycin D to 10ml of ethanol. Store at -20°C.
- Homogenization buffer: 0.375M sorbitol and 20mM HEPES (pH 7.4). To make 500 ml, add 34.2 g sorbitol and 10 ml of 1M HEPES-KOH (pH 7.4) stock to distilled water. Store at 4°C.
- 0.21M KOAc buffer: 0.21M KOAc, 3 mM Mg(OAc)2, and 20mM HEPES (pH 7.4). To make 100ml, add 5.25 ml of 4M KOAc stock, 0.3 ml of 1M Mg(OAc)2 stock, and 2 ml of 1M HEPES-KOH (pH 7.4) stock to distilled water. Store at 4°C.
- Transport buffer: 0.25M sorbitol, 70mM KOAc, 1 mM Mg(OAc)2, and 20mM HEPES (pH 7.4). To make 100ml, add 10ml of 2.5M sorbitol stock, 1.75ml of 4M KOAc stock, 0.1 ml of 1M Mg(OAc)2 stock, and 2ml of 1M HEPES-KOH (pH 7.4) stock to distilled water. Store at -20°C.
- Prepare vesicular stomatitis virus (VSV; Indiana serotype) strain ts045 according to Schwaninger et al. (1992). Store the virus in 1-ml aliquots in screw-capped tubes at -80°C.
- Grow normal rat kidney (NRK) cells on 150-mm tissue culture dishes (Cat. No. 3025; Falcon) in α-MEM medium supplemented with 5% FBS at 37°C and 5% CO2. At confluency, infect the cells with a 5-ml cocktail (per dish) containing 0.1-0.25 ml (2-10 pfu/cell) of ts045 VSV (thawed at 32°C) and 25µg of actinomycin D in serum-free α-MEM as described by Schwaninger et al. (1992). Rock the dishes for 45min to ensure an even spread of the infection cocktail. After infection, add 20ml of α-MEM medium supplemented with 5% FBS to each dish and incubate in the presence of 5% CO2 for 3 h and 40min to 4h at the restrictive temperature (39.5°C) (see Comment 1). The method detailed later is based on a typical 12-dish microsome preparation.
- Following incubation at 39.5°C, transfer each dish to ice, aspirate the medium immediately, and add 12ml of ice-cold Ix PBS to cool the cells as quickly as possible.
- Remove the PBS by aspiration, add 5ml of homogenization buffer, and scrape the cells from the dishes using a rubber policeman. Use a pipette to transfer the cells to 50-ml plastic tubes (Cat. No. 25325- 50: Corning Inc.) and then repeat the scraping procedure to ensure that all the cells are collected. Centrifuge at 720g for 3min and remove the supernatant by aspiration.
- Resuspend each cell pellet (from four dishes) in 0.9ml of homogenization buffer supplemented with PIC and homogenize by three complete passes (three downward strokes with both plungers) through a 1-ml ball-bearing homogenizer (Balch and Rothman, 1985).
- Combine the cell homogenates and dilute with an equal volume (~3 ml) of homogenization buffer + PIC. Divide the diluted homogenate into six 1.0-ml aliquots in 1.5-ml microfuge tubes and centrifuge at 720g for 5 min.
- Carefully remove the postnuclear supernatant (PNS) fractions and combine in a plastic 15-ml tube (Cat. No. 25319-15: Corning Inc.). Add 0.5 volume (~2.5 ml) of 0.21M KOAc buffer to the PNS and mix. Divide the mixture into 0.8- to 1.0-ml aliquots in 1.5-ml microfuge tubes and centrifuge at 12,000g (12,200rpm) for 2min in an Eppendorf Model 5402 refrigerated microfuge using the soft spin function.
- Remove the supernatants by aspiration and resuspend the pellets (including any membranes on the sides of the tubes) using a P1000 Gilson tip in a total volume of 1.0ml of transport buffer + PIC. Dispense 0.5-ml aliquots into two 1.5-ml microfuge tubes and recentrifuge the microsomes at 12,000g for 2min as described earlier.
- Resuspend the membrane pellets by repeated trituration using a Pl000 Gilson tip in 6-8 volumes (~1 ml per 75-µl membrane pellet) of transport buffer containing PIC. In a typical 12-dish preparation, 1.0-1.5ml of resuspended microsomes (at a protein concentration of 3-4mg/ml) is obtained depending on the starting cell density. Pool the membranes, divide into 50- or 100-µl aliquots in 0.5-ml microfuge tubes, freeze in liquid N2, and store at -80°C. The microsomes can be stored for several months with no loss of transport activity.
C. Preparation of Acceptor Golgi Membranes
The following procedure for the preparation of Golgi membranes by flotation on a sucrose density gradient is a modification of that originally described by Balch et al. (1984).
57.5mM KOAc buffer: 87.5 mM KOAc, 1.25 mM Mg(OAc)2, and 20mM HEPES (pH 7.4). To make 100ml, add 2.18ml of 4M KOAc stock, 0.125ml of 1M Mg(OAc)2 stock, and 2ml of 1M HEPES-KOH (pH 7.4) stock to distilled water. Store at 4°C.
Steps
- Prepare an enriched Golgi membrane fraction from noninfected wild-type Chinese hamster ovary cells by flotation in sucrose density gradients as described by Beckers and Rothman (1992). Recover the membranes at the 29-35% sucrose interface, mix thoroughly with 4 volumes of 87.5 mM KOAc buffer, and divide into 0.5- to 1.0-ml aliquots in microfuge tubes. Centrifuge at 16,000 g (soft spin) for 10 min.
- Remove the supernatants by aspiration, wash the pellets with 2 ml (final volume) of transport buffer, and combine the membranes into two 1.5-ml microfuge tubes. Centrifuge at 16,000 g (soft spin) for 10 min.
- Resuspend the membranes using a P1000 Gilson tip in transport buffer (total volume of 1.0ml per 1 × 106 cells). Divide the Golgi membrane fraction into 50- to 100-µl aliquots in 0.5-ml microfuge tubes, freeze in liquid N2, and store at -80°C.
Steps
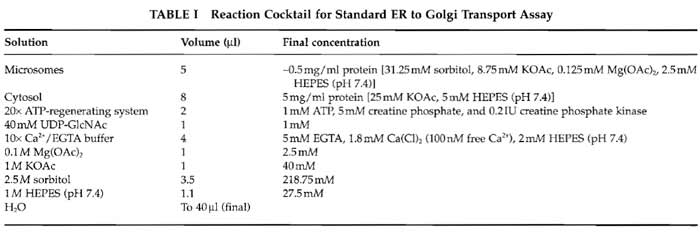
- Set up 40-µl transport reactions containing the components indicated in Table I in 1.5-ml microfuge tubes (see Comment 2). A reaction cocktail consisting of the salts, ATP-regenerating system, and water is added first, followed by the cytosol and finally the microsomes. Mix by pipetting up and down four times using a P20 Gilson tip.
- Transfer the reactions to a 32°C water bath and incubate for 75-90 min.
- Terminate the reactions on ice, harvest the membranes by centrifugation at 20,000g (27,000rpm) in a Beckman TLA 100.3 rotor, and remove the supernatant by aspiration (see Comment 3).
- Solubilize the membranes in one-third volume of 0.3% SDS in 1M Na acetate, pH5.6, and freshly added 30mM BME. Boil 5 min, cool, and two-third volume 1M Na acetate containing 3mU of endo H. Incubate overnight, then terminate the reactions by adding Laemmli sample buffer as described by Schwaninger et al. (1992).
- Separate the endo H-sensitive and -resistant forms of VSV-G on 6.75% (w/v) SDS-polyacrylamide gels as described by Schwaninger et al. (1992).
- Transfer the proteins to a nitrocellulose membrane and perform Western blotting using anti-VSV-G polyclonal primary antibody (1:10,000) and peroxidase- conjugated anti-rabbit IgG secondary antibody (1:10,000) according to Rowe et al. (1996).
- Develop the blots using enhanced chemiluminescence and expose to autoradiography film. Quantitate the relative band intensities of the endo H-sensitive and -resistant forms of VSV-G in each lane by densitometry (see Comments 4 and 5).
 |
E. Vesicle Formation Assay
Solution
Resuspension buffer: 0.25M sucrose and 20 mM HEPES (pH 7.4). To make 100 ml, add 8.55 g of sucrose and 2 ml of 1M HEPES-KOH (pH 7.4) stock to distilled water. Store at 4°C.
- Set up 40-µl reactions as described in Section III,D, step 1, containing 5-10µl of microsomes, 5-12µl of cytosol, salts, and an ATP-regenerating system (see Comment 6).
- Incubate at 32°C for 0-60min and harvest the membranes as described in Section III,D, step 3. The membrane pellets can be stored for several hours at this stage prior to the differential centrifugation procedure described later.
- Add 40 µl of resuspension buffer and disperse the membrane pellets by pipetting up and down 10 times using a P200 Gilson tip (see Comments 3 and 7). Incubate the membranes for 10min on ice and repeat the trituration procedure to resuspend the membranes completely. Add 8.5 µl of a salt mix [7.3 µl of 1M KOAc + 1.2µl of 0.1M Mg(OAc)2] to the resupended membranes and mix by pipetting up and down 5 times with a P20 Gilson tip. Perform the differential centrifugation step at 16,000g (14,000 rpm) for 3 min in an Eppendorf Model 5402 refrigerated microfuge using the soft spin function.
- Using a P200 Gilson tip, carefully take the top 34-µl supernatant fraction from the side of the tube opposite the pellet. Transfer to a 1.5-ml polyallomer microfuge tube and centrifuge at 100,000g (60,000 rpm) for 20min. Carefully aspirate the remaining supernatant fraction from the 16,000-g (medium speed) pellet and the entire supernatant from the 100,000-g (high speed) pellet (see Comment 8).
- Add 50 and 35µl of IX Laemmli sample buffer (Laemmli, 1970) to the medium-speed pellet (MSP) and high-speed pellet (HSP) fractions, respectively, and boil at 95°C for 5min. Determine the relative amounts of VSV-G in the MSP and HSP from each reaction by SDS-PAGE and quantitative immunoblotting as described by Rowe et al. (1996).
- A quicker alternative method to step 2 is as follows: After incubation put the tubes on ice and spin at 16,000g (14,000rpm) for 3min. Carefully remove the top 32µl as described in step 4 and transfer to 1.5-ml polyallomer tubes. Spin at 100,000g (60,000rpm) for 20min. Aspirate supernatant as in step 4 and add 40 and 32µl 1× Laemmli sample buffer to the mediumspeed pellet (MSP) and high-speed pellet (HSP) fractions, respectively. Vortex carefully and boil at 95°C for 5 min.
F. Two-Stage Fusion Assay
Solution
0.25 M sorbitol buffer: 0.25 M sorbitol and 20 mM HEPES (pH 7.4). To make 100 ml, add 10 ml of 2.5 M sorbitol stock and 2ml of 1M HEPES-KOH (pH 7.4) stock to distilled water. Store at 4°C.
- For stage 1 incubations, prepare scaled-up (100µl) vesicle formation reactions containing 25µl of microsomes and 30B1 of cytosol as described in Section IIIE, step 1. Incubate for 10min at 32°C.
- Terminate the reactions by transfer to ice and sediment the membranes as described in Section IIID, step 3.
- Resuspend the membranes in 90 µl of resuspension buffer, add 9.8 µl of salt mix [7.3 µl of 2M KOAc + 2.5 µl of 0.1M Mg(OAc)2], and perform the differential centrifugation step as described in Section IIIE, step 3.
- Withdraw the top 75-ml medium-speed supernatant fraction and recover the vesicles by centrifugation at high speed as described in Section IIIE, step 4.
- Resuspend the HSPs in 25µl of 0.25M sorbitol buffer by pipetting up and down 10 times with a P200 Gilson tip and then add 1.9µl of 1M KOAc (70mM KOAc final concentration) (see Comment 9).
- Set up 40-µl stage 2 reactions containing 10µl of resuspended HSP fraction, 4µl of Golgi membranes, 8µl of cytosol, and an ATP-regenerating system and salts at the final concentrations indicated in Table I (see Comment 6).
- Incubate for 60min at 32°C and terminate the reactions on ice. Harvest the membranes and quantitate the conversion of VSV-G to the endo H-resistant form as described in Section IIID, steps 3-7.
- To reproduce the temperature-sensitive phenotype of ts045 VSV-G transport in vitro, it is necessary to supplement the postinfection medium and homogenization buffer with dithiothreitol as described by Aridor et al. (1996).
- Preparations of the ATP-regenerating system and Ca2+/EGTA buffer are described by Schwaninger et al. (1992). Transport inhibitors such as antibodies are dialyzed against 25/125 prior to addition to the assay, and the volumes of the salts in the reaction cocktail are adjusted to achieve the final concentrations described in Table I. The reactions can be preincubated on ice for up to 45 min with no loss of transport activity.
- All of the membrane-bound VSV-G is sedimented at 20,000g following the 32°C incubation. In the vesicle formation assay, membranes are resuspended in sucrose buffer prior to differential centrifugation.
- The detection system is linear over the range of VSV-G concentrations tested.
- As an alternative to densitometry, VSV-G bands can be detected by direct fluorescence imaging (e.g., using a Bio-Rad GS-363 molecular imaging system).
- In the vesicle formation and two-stage assays, the volumes of salts (see Table I) added to the reaction cocktail are adjusted to account for the salts present in the cytosol and membrane preparations in the assay. UDP-GlcNAc is omitted from the vesicle formation assay and from stage 1 of the two-stage assay.
- Although in the original description of the assay (Rowe et al., 1996) a 0.25 M sorbitol buffer was used to resuspend the membranes prior to differential centrifugation, we have subsequently found that resuspension in 0.25 M sucrose buffer gives higher yields of vesicles at this step.
- Because the HSP is small and translucent, care should be taken to avoid losing it during aspiration of the high-speed supernatant.
- In a typical experiment, multiple stage 1 reactions are performed and the HSPs are resuspended successively in the desired final volume of sorbitol buffer and are then adjusted to 70 mM KOAc.
This work was supported by postdoctoral fellowships from the Cystic Fibrosis Foundation to XW and grants from the National Institutes of Health (GM 42336; GM33301) to WEB.
References
Aridor, M., and Balch, W. E. (1996). Membrane fusion: Timing is everything. Nature 383, 220-221.
Aridor, M., and Balch, W. E. (1999). Integration of endoplasmic reticulum signaling in health and disease. Nature Med. 5, 745-751.
Aridor, M., and Balch, W. E. (2000). Kinase signaling initiates coat complex II (COPII) recruitment and export from the mammalian endoplasmic reticulum. J. Biol Chem. 275, 35673-35676.
Aridor, M., Bannykh, S. I., Rowe, T., and Balch, W. E. (1995). Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 131, 875-893.
Aridor, M., Bannykh, S. I., Rowe, T., and Balch, W. E. (1999). Cargo can modulate COPII vesicle formation from the endoplasmic reticulum. J. Biol Chem. 274, 4389-4399.
Aridor, M., Fish, K. N., Bannykh, G. I., Weissman, J., Roberts, H. R., Lippincott-Schwartz, J., and Balch, W. E. (2001). The Sarl GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J. Cell Biol. 152, 213-229.
Balch, W. E., Dunphy, W. G., Braell, W. A., and Rothman, W. E. (1984). Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell 39, 405-416.
Balch, W. E., and Rothman, J. E. (1985). Characterization of protein transport between successive compartments of the Golgi apparatus: Asymmetric properties of donor and acceptor activities in a cell-free system. Arch. Biochem. Biophys. 240, 413-425.
Beckers, C. J. M., and Rothman, J. E. (1992). Transport between Golgi cisternae. Methods Enzymology 219, 5-12.
Davidson, H. W., McGowan, C. H., and Balch, W. E. (1992). Evidence for the regulation of exocytic transport by protein phosphorylation. J. Cell Biol. 116, 1343-1355.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685.
Lafay, E (1974). Envelope viruses of vesicular stomatitis virus: Effect of temperature-sensitive mutations in complementation groups III and V. J. Virol. 14, 1220-1228.
Rowe, T., Aridor, M., McCaffery, J. M., Plutner, H., Nuoffer, C., and Balch, W. E. (1996). COPH vesicles derived from mammalian ER microsomes recruit COPI. J. Cell Biol. 135, 895-911.
Schwaninger, R., Beckers, C. J. M., and Balch, W. E. (1991). Sequential transport of protein between the endoplasmic reticulum and successive Golgi compartments in semi-intact cells. J. Biol. Chem. 266, 13055-13063.
Schwaninger, R., Plutner, H., Davidson, H. W., Find, S., and Balch, W. E. (1992). Transport of protein between the endoplasmic reticulum and Golgi compartments in semi-intact cells. Methods Enzymology 219, 110-124.
Support our developers




